Chemical Classes of Proteasome Inhibitors in Clinical Development
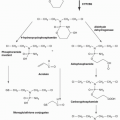
As of the writing of this overview, six different proteasome inhibitors comprising three distinct chemical classes have been tested in clinical trials (
Table 24.1) and include: (1) dipeptide boronic acids, (2) peptide epoxy ketones, and (3) β-lactones.
18,19 Bortezomib (PS-341, Velcade), a dipeptide boronic acid, was developed by Millennium Pharmaceuticals (Cambridge, MA) and was the first PI approved for clinical use.
20 Two additional dipeptide boronic acids have entered clinical development, ixazomib/MLN 9708 (Millennium), currently in phase III studies, and delanzomib/CEP-18770 (Teva Pharmaceuticals; Frazer, PA), the clinical development of which has been halted. Carfilzomib (Onyx Pharmaceuticals; San Francisco, CA), a tetrapeptide epoxy ketone, received U.S. Food and Drug Administration (FDA) approval in 2012.
21 A second peptide epoxy ketone proteasome inhibitor, oprozomib (Onyx), entered clinical study in 2010. The third class of proteasome inhibitors, β-lactones, is represented by NPI-0052 (salinosporamide A [Marizomib]) and is currently being developed by Nereus Pharmaceuticals, Inc. (San Diego, CA). The initial approvals for both bortezomib and carfilzomib were in multiple myeloma (MM), a plasma cell neoplasm and the second most common hematologic cancer. However, the activity of PIs in other B-cell neoplasms has resulted in an expansion of the clinical utilization of this drug class.
Preclinical Activity of Proteasome Inhibitors
Each of the three classes of inhibitors has a distinct chemical mechanism of proteasome inhibition.
22 Peptide boronates form stable but reversible tetrahedral intermediates with the γ-hydroxyl (γ-OH) group of the catalytic N-terminal threonine of the proteasome active sites.
23,24 β-lactones also interact with this γ-OH, but form a completely irreversible interaction.
25 Similarly, peptide epoxy ketones form irreversible covalent adducts with the active site threonine but do so via a dual covalent adduction of γ-OH group and the free amine.
26 This interaction is highly specific for N-terminal threonine-containing hydrolases and renders peptide epoxy ketones the most selective proteasome inhibitors yet described.
27,28The primary targets of these PIs within the constitutive and immunoproteasomes are the CT-L subunits, β5 and LMP7, respectively. Despite accounting for less than 50% of total protein turnover by the proteasome, these subunits are essential for cell survival.
29 In MM cell lines, inhibiting both subunits (β5 and LMP7) is necessary and sufficient for tumor cell death.
30 Cytotoxicity of other tumor cell types requires the inhibition of multiple active sites beyond the CT-L activity. The combination of inhibitors specific for either the T-L or C-L activities, which have no cytotoxic activity on their own, augments the cytotoxic potential of the CT-L-specific inhibitors.
31,32Given its status as the first proteasome inhibitor approved for marketed use, the antitumor potential and preclinical activity of other proteasome inhibitors have generally been compared to bortezomib.
19 Carfilzomib showed equivalent antitumor activity to bortezomib in vitro against a panel of tumor cell lines under standard culture conditions but was >10-fold more potent at inducing tumor cell death when cells were exposed to drug for a 1-hour pulse, which mimics the pharmacokinetics of both compounds.
33 MLN2238 (the active agent of ixazomib) was active in the same mouse models of human tumors as bortezomib, but demonstrated greater levels of proteasome inhibition in the tumors.
34 In biochemical assays of proteasome activity, delanzomib had an identical potency and subunit activity profile to bortezomib, but in tumor cytotoxicity assays, potency relative to bortezomib was 2- to 10-fold less.
35 In addition, delanzomib appeared to be less cytotoxic than bortezomib to normal cells and had a differential effect on cytokine release in bone marrow stromal cells, suggesting a different pharmacologic activity. Oprozomib is 10-fold less potent than carfilzomib in proteasome activity assays, but showed similar antitumor activity in mouse tumor models.
36,37 Marizomib displayed greater potency against the non-CT-L active sites of the proteasome than bortezomib.
38 Interestingly, this agent synergized with bortezomib in killing tumor cells in vitro.
39 All of the second-generation inhibitors have shown activity in tumor cells made resistant to bortezomib and/or MM cells isolated from patients relapsed from bortezomib-based therapies
35,36,40,41,42The inhibition of tumor cells with proteasome inhibitors induces cell death via the induction of apoptosis through death effector caspase activation.
10 Although the mechanism underlying the induction of cell death remains to be fully elucidated, extensive research suggests a complex interplay of multiple pathways. PIs have been shown to affect the half-life of the
BH3-only members of the Bcl-2 family, specifically BH3-interacting-domain death agonist (Bid) and Bcl-2 interacting killer (Bik).
43 Moreover the BH3-only protein NOXA is upregulated at the transcription level by PIs.
44,45,46,47,48 Proteasome inhibition also upregulates the expression of several key cell-cycle checkpoint proteins that include p53 (an inducer of G0/G1 cell-cycle arrest through accumulation of the cyclin-dependent kinase [CDK] inhibitor p27); the CDK inhibitor p21; mammalian cyclins A, B, D, and E; and transcription factors E2F and Rb.
49,50 The transcription factor nuclear factor kappa B (NF-κB), an important regulator of cell survival and cytokine/growth factor production,
51 is also affected by proteasome inhibition in multiple ways. The net effect on NF-κB signaling is not consistent across various assays and cell lines, and its relative importance in the antitumor effects of PIs remains unclear. Although it is interesting to note that patients whose myeloma harbor NF-κB-activating mutations (˜20%) respond better to bortezomib than those without NF-κB-activating mutations.
52,53,54 In MM cell lines, there is growing evidence that the major determinant of sensitivity to proteasome inhibition is the relative load of protein flux to the proteasome.
55,56,57 These data suggest that induction of the terminal unfolded protein response may drive cell death. Whether proteotoxic stress induced cell death reflects sensitivity to proteasome inhibitors in other tumor types remains to be determined.