Programmed Cell Death
Andrew L. Snow
Michael J. Lenardo
INTRODUCTION
Nontransformed cells have a finite lifespan. After a characteristic number of divisions, cells generally undergo senescence, stop dividing, and subsequently die. Considering adult vertebrates, and their internal organs, generally stay a constant size, a form of homeostasis is required.1 In certain organs, this involves a constantly fluctuating dynamic equilibrium of cell proliferation and death. A prime example is the immune system, which employs cell renewal, expansion, and elimination in carrying out its functions.2,3,4,5 These changes can be systemic or localized to anatomic sites proximate to an antigen stimulus, and may affect specific subsets of immune cells as dictated by the conditions.
Programmed cell death (PCD) denotes a set of internal biochemical mechanisms that cause specific cells to die under defined conditions that are typically advantageous to the organism.6 Reasons for cell elimination include cell excess, improper cell differentiation, cell transformation, genetic damage to cells, and infection. In addition to this operational definition, considering a death event “programmed” usually means that one or more genes are required. This chapter has been organized to start with a broad overview of general immunoregulatory principles and then cover details of the molecules involved in PCD in specific sections. Although there are conceptual antecedents from the 19th century, the selection of cells for survival or death by specific external stimuli was introduced by Levi-Montalcini for neural cells in the 1940s and later by Burnet for lymphocytes. Investigation into the molecular mechanisms of PCD began in the mid-1980s and accelerated rapidly. Deoxyribonucleic acid (DNA) sequence databases permitted the rapid identification molecules involved in PCD.7 Although PCD is a large and contentious area of cell biology research, molecular advances have established a firm and tractable theoretical foundation. Remarkably, much of what we will discuss was almost completely unknown two decades ago. Yet these pathways are at work every day in our bodies to control responses to infectious agents, establish cellular homeostasis, prevent autoimmunity, and avert lymphoid malignancies.
OVERVIEW: CELLULAR HOMEOSTASIS AND PROGRAMMED CELL DEATH IN MULTICELLULAR ORGANISMS
Internal programs of death exist in all mammalian cells that likely require constant and active suppression.8 For experimental investigation, it is thus important to discriminate between accidental cell death and that initiated or “programmed” by an internal genetically determined biochemical mechanism. The term “apoptosis,” a Greek word meaning “falling off,” as in leaves from a tree, was introduced in 1972 by Andrew Wyllie and coworkers to describe the normal, presumably programmed, attrition of cells.9 It was defined microscopically as cell shrinkage with nuclear and cytoplasmic condensation within a cell membrane that forms blebs but stays intact until late in the death process10 (Fig. 30.1). This cell phenotype has long been associated with cell death.11 Apoptosis is now mainly identified by the biochemical effects of the caspase family of proteases.12 Caspases are important in two respects. First, they are a feature of most, if not all, apoptosis pathways. Second, once highly activated, they usually represent a commitment to apoptosis that is not reversible, although we now recognize that low-level caspase activation participates in lymphocyte activation.13,14 For these reasons, caspases have been regarded as a final common pathway of apoptosis. In fact, the concept of PCD was significantly illuminated by the identification of caspases and other molecules that regulate these internal biochemical death pathways. In general, the molecular components of the death mechanism are preassembled and available without new gene transcription or protein synthesis.6 For example, caspases are constitutively expressed in the cytoplasm of the cell as zymogens. Once proteolytically activated, caspases carry out specific protein cleavages leading to the morphologic changes of apoptosis. The proteolytic substrates of caspases are highly selective because most proteins remain uncleaved. A panoply of molecular events entrained to caspases includes cleavage of chromosomal DNA, nuclear chromatin condensation, exposure of phosphatidylserine on the exterior of the cell membrane, proteolysis of specific proteins including other caspases, and mitochondrial changes. These processes are detectable by simple assays in tissue culture cells in vitro or, in some cases, in vivo. Protocols for these assays have been well described.15,16
Cells that die without the characteristics of apoptosis typically undergo what is usually called necrosis. While necrosis is often associated with accidental cell death such as physical or chemical trauma, it may also result from programmed mechanisms. In certain cases, nonapoptotic mechanisms of cell death can be promoted by caspase inhibition.17 The appearance of a necrotic cell differs dramatically from that of an apoptotic cell. Necrotic cells swell and lose the integrity of internal organelles with early plasma
membrane breakdown, giving an enlarged “fractured” appearance under the microscope (see Fig. 30.1D). Recently, various research groups have begun to define molecular programs resulting in necrotic death.18 Several varieties of “programmed” necrosis that share the hallmarks of early loss of plasma membrane integrity and excessive reactive oxygen species (ROS) have been described. Programmed necrosis that involves receptor-interacting protein (RIP) serine/threonine kinases has been well documented in lymphocytes, especially under conditions of caspase inhibition.17,18 A closely related variant of this cell death program termed “necroptosis” is inhibitable by necrostatin, a compound that inhibits RIP kinases.19 Finally, the induction of autophagy has been genetically linked to a necrotic program of death.20 Autophagy is induced by starvation or other stimuli and involves internal membrane rearrangement leading to engulfment of portions of the cytoplasm, which are then degraded by fusion with lysosomes. Under death conditions, this mechanism leads to membrane damage and necrosis by ROS, which overaccumulate because of accelerated catalase degradation.21 The precise role of these necrosis programs, especially autophagic cell death, in immune function has not yet been worked out. However, they are often provoked by the inhibition of caspase-dependent apoptosis. Caspase inhibition, perhaps by blocking constitutive low-level caspase activity, can actually trigger these necrotic death programs.20
membrane breakdown, giving an enlarged “fractured” appearance under the microscope (see Fig. 30.1D). Recently, various research groups have begun to define molecular programs resulting in necrotic death.18 Several varieties of “programmed” necrosis that share the hallmarks of early loss of plasma membrane integrity and excessive reactive oxygen species (ROS) have been described. Programmed necrosis that involves receptor-interacting protein (RIP) serine/threonine kinases has been well documented in lymphocytes, especially under conditions of caspase inhibition.17,18 A closely related variant of this cell death program termed “necroptosis” is inhibitable by necrostatin, a compound that inhibits RIP kinases.19 Finally, the induction of autophagy has been genetically linked to a necrotic program of death.20 Autophagy is induced by starvation or other stimuli and involves internal membrane rearrangement leading to engulfment of portions of the cytoplasm, which are then degraded by fusion with lysosomes. Under death conditions, this mechanism leads to membrane damage and necrosis by ROS, which overaccumulate because of accelerated catalase degradation.21 The precise role of these necrosis programs, especially autophagic cell death, in immune function has not yet been worked out. However, they are often provoked by the inhibition of caspase-dependent apoptosis. Caspase inhibition, perhaps by blocking constitutive low-level caspase activity, can actually trigger these necrotic death programs.20
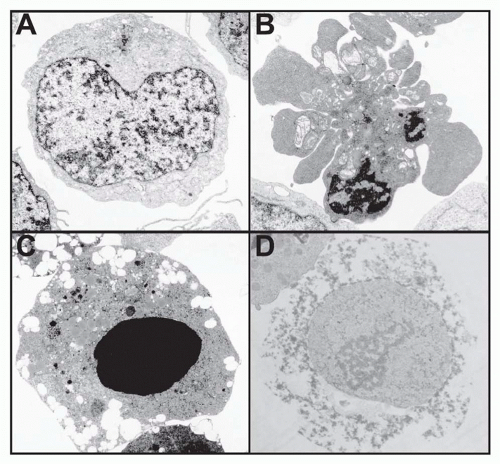 FIG. 30.1. Electron Microscopy Elucidates the Morphology of Different Forms of Programmed Cell Death. A: A normal, unstimulated Jurkat leukemia cell. B: Jurkat cell undergoing apoptosis in response to Fas receptor stimulation. Notice the condensed chromatin and the blebbing with maintenance of plasma membrane integrity. C: Jurkat cell undergoing apoptosis in response to staurosporine treatment. Note the prominent nuclear condensation. D: Necrosis of human peripheral blood T-lymphocyte infected with the human immunodeficiency virus. Notice the general loss of cellular integrity as well as the lack of chromatin condensation in the dying cell. (Figure courtesy of Diane Bolton and Jan Orenstein.) |
Thus, it is important to distinguish between “programmed” necrosis and necrosis due to accidental causes as these involve very different molecular events. It has been generally argued that apoptosis, which preserves membrane integrity, prevents inflammation from released cellular contents, whereas necrosis results in total cellular breakdown and content release, which causes an inflammatory response.22 Recent evidence suggests this paradigm is oversimplified and that the immunogenicity of a dying cell varies with cell type and activation status, the nature of the phagocytic cell that engulfs it, and/or the specific death stimulus. The release of inflammatory damage-associated molecular patterns that may accompany apoptosis or necrosis of stressed cells (eg, heat shock proteins, high-mobility group box 1, oligonucleotides, uric acid) may be particularly important for invoking a strong immune response.23 These distinct immunologic effects due to apoptosis and necrosis remain the subject of continued experimental exploration.
As we will argue in the following, the necessity of cellular homeostasis as well as the acute need to eliminate cells that are harmful or nonfunctional led to the early emergence of conserved cell death mechanisms during evolution.11. Work by Horvitz and colleagues genetically identified several molecules essential for the death of specific cells during the development of the roundworm Caenorhabditis elegans that have subsequently been found to be homologues for mammalian PCD genes.24 It is clear from this simplified system that the molecular logic of one form of cell death was likely established early in evolving multicellular organisms.25 PCD mechanisms are now evident in most contemporary multicellular organisms from plants to humans, although it is not clear whether convergent evolution or conservation of function is responsible. However, the molecular pathways in worms and other simple organisms are rudimentary compared with the complexity found in mammalian PCD systems. To understand how these mechanisms contribute to immunity, we focus on mice and humans, which are the subjects of most immunologic research. Also, while we focus mainly on research carried out on B- and T-lymphocytes, similar themes and mechanisms govern death programs for other hematopoietic cells including dendritic cells (DCs), macrophages, natural killer cells, granulocytes, and mast cells, among others.
PROGRAMMED CELL DEATH AND IMMUNE REGULATION
The fundamental unit of immune responsiveness is the cell. Lymphocytes, the major adaptive immune cells, are distinguished by expressing a unique clonotypic antigen receptor. A large number of lymphocytes with different receptor specificities are generated during ontogeny, providing a diverse “immune repertoire,” and these cells can be programmed to expand or die throughout the life of the organism. Somatic hypermutation during B-cell expansion in the germinal center can expand the repertoire. Although the level of antigen presentation, the degree of lymphocyte responsiveness (versus unresponsiveness or anergy), and other factors also play important roles, the presence or absence of cells with
specific recognition properties at any given time is a primary determinant of the quantitative response to any antigenic stimulus. The homeostasis of major lymphocyte populations is independently regulated such that deficits in B cells, T cells, or even major T-cell subpopulations (&agr;&bgr; versus &ggr;&dgr; or cluster of differentiation [CD]4 versus CD8) may qualitatively alter but do not prevent the normal homeostasis of the remaining populations. During thymic development, lymphocytes respond to the antigenic environment with either survival or death.26 Developmental PCD eliminates lymphocytes that cannot recognize antigen appropriately or have potentially dangerous self-reactivity.26,27 In the mature immune system, death is principally a negative feedback response that counterbalances proliferative responses to antigen.28 Although the clonal selection theory of F.M. Burnet encompassed clonal elimination during ontogeny, it allowed only selective expansion of antigen-stimulated lymphocytes in the mature immune system. However, antigen-specific regulation of mature lymphocyte survival also powerfully controls immune responses and tolerance.4,28,29 Because the organism encounters an unpredictable universe of antigens in a lifetime, it is essential that there is feedback regulation of adaptive immune responses. Feedback is an essential element of any dynamic system in which final outcomes cannot be predicted from the starting conditions.30 To achieve measured immune responses, proliferation and death are coordinated by feedback regulation to control the number of responsive immune cells. Feedback death mechanisms can also eliminate potentially harmful specificities that increase unexpectedly during immune reactions. Hence, homeostasis of both lymphocyte numbers and reactivities can be continuously maintained.
specific recognition properties at any given time is a primary determinant of the quantitative response to any antigenic stimulus. The homeostasis of major lymphocyte populations is independently regulated such that deficits in B cells, T cells, or even major T-cell subpopulations (&agr;&bgr; versus &ggr;&dgr; or cluster of differentiation [CD]4 versus CD8) may qualitatively alter but do not prevent the normal homeostasis of the remaining populations. During thymic development, lymphocytes respond to the antigenic environment with either survival or death.26 Developmental PCD eliminates lymphocytes that cannot recognize antigen appropriately or have potentially dangerous self-reactivity.26,27 In the mature immune system, death is principally a negative feedback response that counterbalances proliferative responses to antigen.28 Although the clonal selection theory of F.M. Burnet encompassed clonal elimination during ontogeny, it allowed only selective expansion of antigen-stimulated lymphocytes in the mature immune system. However, antigen-specific regulation of mature lymphocyte survival also powerfully controls immune responses and tolerance.4,28,29 Because the organism encounters an unpredictable universe of antigens in a lifetime, it is essential that there is feedback regulation of adaptive immune responses. Feedback is an essential element of any dynamic system in which final outcomes cannot be predicted from the starting conditions.30 To achieve measured immune responses, proliferation and death are coordinated by feedback regulation to control the number of responsive immune cells. Feedback death mechanisms can also eliminate potentially harmful specificities that increase unexpectedly during immune reactions. Hence, homeostasis of both lymphocyte numbers and reactivities can be continuously maintained.
Thymic Deletion: Positive and Negative Selection
Thymic selection represents an intriguing example of apoptosis in which the same receptor—the clonotypic T-cell receptor (TCR)—can lead to diametrically opposite outcomes depending on the level of stimulation.26,27 During development, when thymocytes (ie, T-cell progenitors in the thymus) express the TCR and both the CD4 and CD8 coreceptors (the “double positive” stage), thymocytes will undergo apoptosis if they receive no TCR stimulation. This process, called “death by neglect,” will eliminate thymocytes that have not productively rearranged the TCR genes or have no capacity to recognize antigen in the context of self-major histocompatibility complex.31 “Low-level” stimulation of the TCR antagonizes death by neglect. This protective event insures major histocompatibility complex-specific antigen recognition by T cells and is called positive selection.32 While weak TCR signals can deliver an antiapoptotic stimulus, strong TCR engagement of double positive thymocytes delivers a proapoptotic signal. This event, termed negative selection, prevents the emergence of strongly autoreactive lymphocytes from the thymus.31 This deletion step is a major mechanism of central tolerance and the prevention of autoimmunity.33 These processes of selection employ caspase-dependent apoptosis and rapid phagocytosis of the dead thymocytes.34,35 Hence, thymocytes travel a narrow bridge of TCR avidity during development and will die if they deviate from it.
The differences between the neglect (or null), weak, and strong signals that result in life or death appear to be determined at early stages of TCR signaling.36,37 Death receptors (DRs; see the following discussion) appear not to be crucial; instead, there is a direct connection of the TCR signaling apparatus to mitochondrial death pathways.38,39,40,41 Though there is not complete certainty how the TCR dictates life or death at specific antigen levels, the answer to this puzzle will almost certainly reside in the complex signal pathways emanating from this receptor. TCR engagement that causes transient induction of the Erk kinase is associated with positive selection, whereas slow but constant Erk activity is associated with negative selection.42,43,44 Other distinctions in TCR signaling have been identified. Signaling through phosphotidylinositol-3 kinase, the antiapoptotic Akt kinase, and the retinoid orphan receptor-gamma may promote thymocyte survival.36,45 Genetically modified mice have revealed that several transcription factors, such as E2A, Nur77, Id3, and IRF-1, can affect thymic cellularity, indicating that differential signaling may trigger transcriptional events that regulate cell survival.46,47,48,49,50 Finally, Andreas Strasser has emphasized that various forms of physiologic cell death are likely to involve the subset of BH3-only proteins in the Bcl-2 family.51,52 The activation of proapoptotic BH3-only proteins, such as Bak, Bid, and especially Bim, by TCR signals initiates mitochondrial apoptosis during thymic selection processes. Thus, early TCR-induced signaling differences directly entrain transcriptional events that modulate BH3-only regulators of mitochondrial apoptosis. These distinct signals are tied to specific microenvironments within the thymus in which positive and negative selection occur.53 Hence, by intracellular communication with distinct apoptosis regulatory molecules, the TCR has a remarkable ability to signal life or death by the apparent strength of stimulus it receives.
Programmed Cell Death and the Homeostasis of Peripheral T Cells
Death of peripheral T cells differs markedly from thymocyte selection in that PCD of mature lymphocytes occurs mainly in cells that have proven usefulness (ie, they have been already selected in the thymus and activated by antigen). This is because PCD of mature T cells is employed primarily to counter antigen-driven proliferation of activated T cells, including those that could be autoreactive. In general, most naïve lymphocytes survive and circulate in the body in a resting state (G0). The survival of such resting T cells is constitutively maintained by the presence of contact with major histocompatibility complex, the cytokine interleukin (IL)-7, and expression of the antiapoptotic protein Bcl-2.33,54,55,56 During an active immune response, T-lymphocyte proliferation can involve as much as a 10,000-fold expansion within days. Such explosive proliferation is necessitated by the extraordinarily rapid propagation of microbial pathogens. However, these activated and cycling T cells are potentially damaging due to toxic effector functions and potential
cross-reactivity with self-antigens. Activated T-cell expansion does not go unchecked and is subject to negative feedback in the form of cell death. However, because immune responses are directed at specific antigens, they must be independently regulated because some responses might expand while others contract. The immune system has developed propriocidal mechanisms to control independent populations of activated T cells. Propriocidal regulation refers to the various negative feedback death mechanisms that maintain homeostasis of mature peripheral T cells in an antigen-specific manner. These potently restrain antigen-activated T cells and tightly control lymphocyte numbers during and at the conclusion of immune responses.
cross-reactivity with self-antigens. Activated T-cell expansion does not go unchecked and is subject to negative feedback in the form of cell death. However, because immune responses are directed at specific antigens, they must be independently regulated because some responses might expand while others contract. The immune system has developed propriocidal mechanisms to control independent populations of activated T cells. Propriocidal regulation refers to the various negative feedback death mechanisms that maintain homeostasis of mature peripheral T cells in an antigen-specific manner. These potently restrain antigen-activated T cells and tightly control lymphocyte numbers during and at the conclusion of immune responses.
Propriocidal regulation of T cells is triggered by remarkably simple attributes of T-cell activation: the level of cell cycling and the level of antigen restimulation.28,54 These two features are ideally suited for triggering negative feedback death because they provide both a “sensing” mechanism for the level of active T-cell proliferation and a negative response mechanism sensitive to the level of antigen stimulation. There are essentially two different mechanisms: 1) active or antigen-stimulated PCD and 2) passive or lymphokine withdrawal PCD (Fig. 30.2).4,57,58 These have different roles and occur at different times, as will be described in detail in the following. In some respects, it is paradoxical that lymphocytes that respond well to foreign antigen and presumably could have protective value are actively eliminated. However, it is vital to constrain the number of activated T cells to prevent unhealthy effector or autoimmune reactions. During a robust immune response, lymphocytes that cross-react with self-antigens may also proliferate. The propriocidal mechanisms that cause these cells to die upon encountering self-antigens could be an important mechanism of preserving self-tolerance. By this formulation, tolerance is a quantitative effect that is due to the low number of significantly self-reactive lymphocytes in the naïve organism. Clonal expansion during immune responses can unleash dormant or infrequent self-reactive clones, called “forbidden clones” by Burnet, creating an autoimmune diathesis. Propriocidal death reduces these clones and thereby promotes tolerance. Active antigeninduced propriocidal death, which is induced by high or repeated doses of antigen, is especially well suited for the elimination of self-reactive clones because self-antigen is likely to be present in continuous and potentially high amounts.28,54
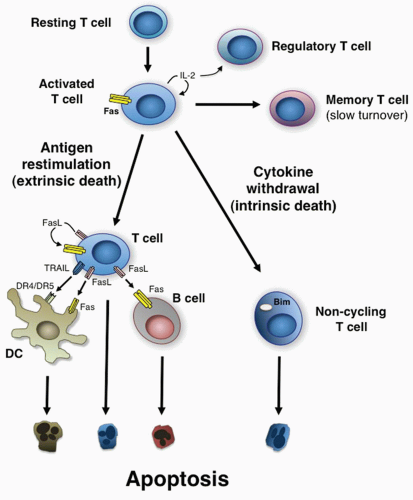 FIG. 30.2. Propriocidal Regulation of Immune Cells. Shown are the apoptosis pathways that govern T-lymphocyte homeostasis by the antigen-restimulated (extrinsic death) and no antigen (cytokine withdrawal [intrinsic death]) pathways of apoptosis. Also shown is the regulation of B cells by FasL expressed by T cells, dendritic cells by Fas ligand/tumor necrosis factor-related apoptosis-inducing ligand expressed by T cells, and T cells by regulatory T cells competing for cytokines. |
Extrinsic or Antigen-stimulated T-Lymphocyte Death
The extrinsic death mechanism involves apoptosis of mature T cells in response to antigen stimulation. This requires the T cells to be activated and cycling at the time that they undergo strong antigenic restimulation.28,54 The death is indirect in the sense that it requires the antigen-induced secretion of death ligands that engage specific apoptosisinducing DRs in the tumor necrosis factor (TNF) receptor (TNFR) and ligand superfamilies (see Fig. 30.2).59,60 Current evidence suggests that Fas ligand (FasL) as well as TNF are the key death ligands that mediate this process in mature CD4+ and CD8+ T cells.4,5,61 However, DR-independent apoptosis mediators also contribute in specific contexts. For example, Th2-differentiated CD4+ cells are less sensitive than their Th1-differentiated counterparts to Fas-mediated killing.62 Instead, they preferentially die through a suicidal pathway that triggers internal release of granzyme B.63,64 Nagata originally observed that the lymphoproliferative and autoimmune phenotype of lpr mice was due to genetic alterations of Fas and a similar disease was due to a mutation in FasL.65,66 Defects in Fas (CD95) or FasL in mice and humans cause severe derangements of lymphocyte homeostasis and tolerance, which will be detailed subsequently. However, activated Fas-deficient T cells can be induced to die through other apoptotic signals, including cytotoxic granules containing perforin and granzymes, and/or TCR-induced upregulation of Bim. These alternative mechanisms may be particularly relevant to clearance of activated CD8+ T cells.67,68,69,70,71,72,73 Often, these forms of death are called “activation-induced cell death,” but this is a misnomer.74 Activation-induced cell death has been used to describe any form of death of activated T cells, thus causing confusion among investigators working on molecularly distinct death pathways.4,75 Activation per se
does not directly cause cell death; instead, extrinsic death induction requires antigenic restimulation of activated T cells (ie, TCR reengagement after initial activation). For resting T cells, antigen encounter under costimulatory conditions leads to activation with very little cell death. Obviously, if the initial activation directly induced death, this would preclude immune responses. On the contrary, activated, cycling T cells do not spontaneously die by FasL or TNF unless they are strongly restimulated by antigen in the activated state, which causes upregulation of the genes for these death cytokines and responsiveness to them.76 For these reasons, we prefer to use the term “restimulation-induced cell death” (RICD) to clarify this important distinction.77,78
does not directly cause cell death; instead, extrinsic death induction requires antigenic restimulation of activated T cells (ie, TCR reengagement after initial activation). For resting T cells, antigen encounter under costimulatory conditions leads to activation with very little cell death. Obviously, if the initial activation directly induced death, this would preclude immune responses. On the contrary, activated, cycling T cells do not spontaneously die by FasL or TNF unless they are strongly restimulated by antigen in the activated state, which causes upregulation of the genes for these death cytokines and responsiveness to them.76 For these reasons, we prefer to use the term “restimulation-induced cell death” (RICD) to clarify this important distinction.77,78
Extrinsic death is a negative feedback mechanism, which explains why it is triggered by lymphokine-induced cell cycling (usually caused by IL-2) and reengagement of the TCR. IL-2-induced cell cycling indicates that there has been a productive antigen response and the T cells are multiplying. Because T cells can expand rapidly to great numbers, a large fraction of cycling T cells dictates a need to downregulate the response to any further antigen exposure. The presence of repeated or continuously high amounts of antigen would be a powerful stimulus to greater proliferation. Under these conditions, the system programs a fraction of the restimulated cells to undergo apoptosis via RICD. RICD sensitivity is dictated in part by the relative strength of the TCR restimulation signal, which must meet a certain “threshold” to fully activate proximal signaling proteins like CD3&zgr; and induce sufficient expression of proapoptotic molecules.79,80 It remains unclear how this apoptosis threshold is calibrated in any given T cell, although additional signals provided through certain signaling lymphocyte activation molecule receptors and the small SH2 adaptor signaling lymphocytic activation molecule-associated protein appear critical for potentiating TCR signal strength in RICD.81 Another key determinant of death in this context is T-cell phenotype. Specifically, among CD4+ T cells, those with an effector memory phenotype are highly sensitive to Fas- and TCR-induced apoptosis, whereas central memory and activated naïve CD4+ T cells are comparatively resistant.82 The molecular basis of this sensitivity to Fas-induced apoptosis depends on the enrichment of Fas in lipid raft microdomains in the plasma membrane of effector memory T cells, which leads to more effective nucleation of the Fas signaling complex. Thus, Fas-/TCR-induced apoptosis can specifically cull extraneous effector memory T cells without diminishing subsequent immune responses by a simple and specific feedback loop. Like most negative feedback systems, the propriocidal response directly reverses the ongoing process of proliferation by eliminating activated T cells. Antigeninduced death provides an explanation for many historical observations in the literature that show that reapplication or continuous presence of high concentrations of antigen can lead to specific suppression rather than augmentation of an immune response.54
Sensitivity to this extrinsic death mechanism is also directly connected to the effect of IL-2 in inducing cell cycle progression into late G1 or S phase, which confers susceptibility to death.83,84,85 The requirement for cell cycle progression has not been fully explained but appears to be necessary for apoptosis induced by TCR engagement or with direct Fas stimulation. Other cytokines that augment T-cell cycling, such as IL-4, IL-7, and IL-15, can also promote cell death to some degree but none with the potency of IL-2.76 Hence, the theory of propriocidal regulation advanced the concept that IL-2 would have an important regulatory role in the elimination of activated T cells in addition to its previously known role in lymphocyte proliferation.54 This concept was later validated when genetic deficiencies of IL-2 and IL-2 receptor in mice were found to have defective apoptosis of activated T cells and autoimmunity.86,87 Although the immunopathology in these animals is also attributed to loss of T regulatory (Treg) cells, the latter constitutes another form of polyclonal deletion by cytokine competition, which we explain in detail subsequently. This surprising property of IL-2 is important to consider in the use of IL-2 as a therapeutic agent or vaccine adjuvant. It also underscores an important feature of feedback regulation: that to achieve a maximal response for, say, a vaccine, more stimulation is not necessarily better. In a variety of test situations, extrinsic antigen-induced death decreases the number of T cells but does not completely eliminate the T-cell immune response.28,29 In certain extraordinary conditions, such as high levels of a noncytopathic or chronic virus, essentially all responding T cells can be eliminated.88 FasL expressed on T cells also causes the death of B cells, which do not themselves express FasL.89 This causes a parallel regulation of B-cell proliferation by Fas-induced death that can be antagonized by B-cell receptor engagement (see Fig. 30.2). Antigen-induced death therefore provides a mechanism to eliminate specific antigen-reactive lymphocytes under chronic stimulatory conditions when they might cause the host more harm than good.
Antigen-induced expression of death ligands and other proapoptotic molecules shunts a proportionate fraction of the antigen-specific activated cells, but not bystander cells, into the death pathway. The activated T cells still carry out their effector function when restimulated by antigen, but their ultimate fate is death instead of proliferation.76,88 Because the primary agents of T-cell death, Fas and other TNFRs, have no inherent antigen specificity, it is important to consider how the clonal specificity of antigen-induced apoptosis is achieved.28 For the death of activated T cells, the simple engagement of Fas by its ligand is insufficient.90,91 Efficient death induction also requires a “competency” signal from the TCR delivered at the same time FasL binds Fas.90,92 However, these signals do not require new protein synthesis and are delivered rapidly in a few hours or less.90,91 The requirement for simultaneous engagement of Fas and TCR plays a critical role in establishing the antigen specificity of death. For example, it was shown that TCR stimulation of a specific subpopulation within a pool of Fas-expressing T-cell blasts, such as with agonistic anti-V&bgr;8 antibody, leads only to the death of that subpopulation despite the apparent exposure of other subtypes of T cells to the V&bgr;8-expressing cells that have been induced to express FasL.28,90 Also, deletion in vivo is antigen-specific.93
The molecular nature of the competency signal is presently unknown.
The molecular nature of the competency signal is presently unknown.
Intrinsic Growth Factor Withdrawal T-Cell Death
As important as it is to avert cellular overexpansion during an immune response through extrinsic, TCR-induced death, it is equally important for the immune system to downmodulate the immune reaction after successful elimination of the pathogen. Lymphokine withdrawal death is a form of T-cell apoptosis that occurs naturally at the end of an immune response when the accumulation of effector cells becomes unnecessary and potentially damaging.94 When the trophic cytokine for activated T cells, typically IL-2, decreases because of reduced antigen stimulation, the excess T cells undergo apoptosis.95 This form of negative feedback death may affect specific classes of T cells, for example, “effector” versus “memory” cells, though this distinction may be difficult to discern experimentally. As most T cells in the expanded population are antigen-specific, this represents clonotype-specific T-cell propriocidal regulation controlled by antigen and cell cycle progression. The activated cells can “sense” decreased antigen drive and decreased trophic cytokine, which programs them for apoptosis. The elimination of the expanded pool of activated cells, except for a small amount of memory cells, reestablishes homeostasis in T-cell numbers. It has been shown that if IL-2 is exogenously delivered during a proliferative response to superantigen, the reactive T cells persist as long as lymphokine is provided.96 Therefore, the lack of IL-2 is a key element in the feedback regulation of the cellular response. Antigen and IL-2 therefore mediate propriocidal regulation, in the midst of an immune response and at its conclusion to reduce T-cell numbers.
Genetic studies reveal that the molecular mechanism of lymphokine withdrawal death is different than antigen-induced apoptosis.57,58 Although this event is often confused with extrinsic cell death mediated by Fas, DRs are not involved. Rather, cytokine withdrawal for as little as 2 to 4 hours commits the cell to a death pathway requiring new protein synthesis.97 Apoptosis is initiated through the mitochondrial pathway and is orchestrated by the Bcl-2 family of proteins. In fact, the ratio between the pro- and the antiapoptotic Bcl-2 family members is believed to determine the fate of the cells. In the presence of growth factors, the antiapoptotic Bcl-2, Bcl-XL, and Mcl-1 proteins maintain viability via retrotranslocation of the executioner molecules Bax and Bak from the mitochondria into the cytosol, precluding oligomerization and permeabilization of the mitochondrial outer membrane (see following section).98 Proapoptotic, “BH3-only” activators like Bim and Bid can bind to antiapoptotic Bcl-2 family proteins and disrupt this process to allow Bax-/Bak-mediated mitochondria depolarization. Bim is an essential regulator upstream of Bax and Bak during cytokine withdrawal-induced apoptosis in lymphocytes.99 CD4+ and CD8+ T cells from mice homozygously deficient for Bim survive much longer than their wild-type counterparts following in vitro and in vivo activation with the superantigen Staphylococcus aureus enterotoxin B.100 Moreover, Bim is critical for cell death of T cells induced by both IL-2 and IL-7 withdrawal.51,100,101 Bim dysregulation also underlies the overaccumulation of lymphocytes due to failed cytokine deprivation apoptosis in the lymphoproliferation disorder caused by activating mutations of Ras.102 Cytokine deprivation rapidly upregulates Bim messenger ribonucleic acid levels through activation of the forkheadlike transcription factor FOXO3A. Bim is also subject to sophisticated posttranscriptional regulation in T cells. For example, removal of IL-2 shuts down the Ras-ERK signaling cascade emanating from the IL-2 receptor, which normally promotes Bim protein degradation and destabilization of Bim messenger ribonucleic acid.103,104 PUMA, another BH3-only protein, functions synergistically with Bim downstream of FOXO3A and accounts for Bim-independent death.105,106 When the trophic cytokine is present, the Ser/Thr kinase Akt also phosphorylates key substrates and prevents mitochondrial collapse. For example, Akt suppresses FOXO3A activity. Akt also phosphorylates the pro-apoptotic Bad protein, which is then sequestered by the 14-3-3 scaffold protein.107,108 Another target inactivated by Akt is glycogen synthase kinase-3, which phosphorylates Mcl-1 on residue S159 when cytokines are removed. The rapid degradation of phosphorylated Mcl-1 by the ubiquitin-proteasome pathway disrupts the fragile equilibrium between the pro- and the antiapoptotic Bcl-2 family members, and promotes cell death.109 Thus, Akt can preserve viability by targeting appropriate Bcl-2 family members. Ultimately, the relative balance of proapoptotic (Bim, Puma) versus antiapoptotic (Bcl-2, Mcl-1, etc.) proteins expressed regulates mitochondrial stability and apoptosis sensitivity in response to cytokine withdrawal.
T Regulatory Cells
Research on the intriguing subset of CD4+ T cells known as Treg cells, and their role in immune tolerance, continues to expand vigorously. These cells express the forkhead box P3 transcription factor and suppress the expression of many TCR-induced genes including cytokines and cytokine receptors.110,111 Similar to other subsets of T cells in their “activated” state, Tregs require common gamma chain cytokines for survival. In particular, they characteristically express the IL-2 alpha chain receptor (CD25) and require IL-2 to persist in the periphery.112 IL-7 can also act as a survival factor for Treg cells.113 The mechanisms by which Treg cells “suppress” immune responses have remained somewhat obscure. However, recent evidence suggests that the direct Treg-mediated suppression of other T cells can be attributed in large part to their large appetite for survival cytokines, which Tregs themselves cannot produce.114,115 Tregs absorb significant amounts of trophic cytokines like IL-2 at a site of an immune reaction, which induces Bim-dependent, intrinsic lymphokine withdrawal death of nearby activated and cytokine-requiring T cells.116 Consistent with historical observations, this phenomenon is not antigen-specific but requires Tregs to be in close proximity to IL-2-producing T cells to effectively consume the cytokine and preclude autocrine/paracrine signaling in
targeted responder T cells.115,117 Tregs can also consume other common gamma chain cytokines involved in lymphocyte survival and effector function, such as IL-4, IL-7, IL-15, and IL-21.118 Therefore, this form of Treg-mediated polyclonal deletion constitutes a form of dominant tolerance dependent on intrinsic PCD. This mechanism allows a productive immune response to unfold while the ratio of effector T cells to Tregs remains high. Subsequent IL-2-mediated expansion of Tregs then establishes a natural brake on effector cell expansion via competition for available cytokines and culling of excess effectors deprived of IL-2. As polyclonal deletion is enforced, Tregs may also utilize alternative strategies for functional suppression of remaining responder T cells or antigen-presenting cells, including the production of suppressive cytokines (eg, transforming growth factor-&bgr;, IL-10) and cytotoxic T-lymphocyte antigen-4 effects on costimulatory molecules (CD80, CD86).119
targeted responder T cells.115,117 Tregs can also consume other common gamma chain cytokines involved in lymphocyte survival and effector function, such as IL-4, IL-7, IL-15, and IL-21.118 Therefore, this form of Treg-mediated polyclonal deletion constitutes a form of dominant tolerance dependent on intrinsic PCD. This mechanism allows a productive immune response to unfold while the ratio of effector T cells to Tregs remains high. Subsequent IL-2-mediated expansion of Tregs then establishes a natural brake on effector cell expansion via competition for available cytokines and culling of excess effectors deprived of IL-2. As polyclonal deletion is enforced, Tregs may also utilize alternative strategies for functional suppression of remaining responder T cells or antigen-presenting cells, including the production of suppressive cytokines (eg, transforming growth factor-&bgr;, IL-10) and cytotoxic T-lymphocyte antigen-4 effects on costimulatory molecules (CD80, CD86).119
T-Cell Memory
T cells, once activated, can persist as “memory” cells. One view is that this process involves an escape from propriocidal mechanisms of apoptosis.35,120,121,122 Increasing evidence supports the concept that memory is due to the long-term survival of antigen-specific T cells even without further antigen exposure. Several means to achieve such survival are possible. To escape killing by Fas and other death receptors, T cells could express cellular FLICE inhibitory protein (FLIP), which is a homologue of caspase-8 and -10 located in the same genetic locus as these caspases that has no enzymatic function but can interpose itself into the death receptor complex and block caspase activation.123 This type of inhibition has been demonstrated in B cells by the ability of B-cell receptor (BCR) stimulation, which upregulates cellular FLIP,124,125 to block Fas killing.126 Specialized T-cell subsets, including proinflammatory Th17 cells, may escape Fas-mediated RICD through cellular FLIP overexpression.127 Various “inhibitor of apoptosis proteins” (IAPs) might also interfere with DR-mediated killing.128,129 Furthermore, the mitochondrial death pathway could be attenuated by upregulation of Bcl-2 and Bcl-XL. These antiapoptotic molecules, which are upregulated in CD8+ and CD4+ memory cells,122,130 have been shown to block lymphokine withdrawal apoptosis.131 The necessity of these inhibitory molecules for the persistence of a memory population of T cells is unknown. Moreover, it is mainly the central memory rather than effector memory T cells that become refractory to RICD.82
Another view is that long-term survival in a nonproliferative “resting” state characteristic of “virgin” (naïve) T cells is never reestablished by memory cells. Work by Phillipa Marrack, Rafi Ahmed, and others reveals a surprisingly dynamic T-cell memory pool. Their study proposes a constant, low-level proliferation of memory CD8+ T cells that is antigen-dependent and maintained by IL-15 and to a lesser extent by IL-7.132,133,134 Also, the fraction of memory cells remains constant over time, indicating that death is continuously maintaining a balance. Hence, the balance of slow proliferation and slow death ensures memory cell maintenance. As the molecular mechanism of the memory state is further elucidated, the differing views of “memory” are likely to be reconciled.
The mitochondrial pathway of death is likely to play a prominent role in the generation of memory cells. In addition to their proliferative effects, the cytokines IL-15 and IL-7 appear to promote survival of memory cells through their induction of antiapoptotic molecules such as Bcl-2.135,136,137 This countervailing effect is consistent with a recently demonstrated Bim-dependent mechanism for limiting memory CD8+ T cells.138 In addition, a Bim-independent mechanism for expunging memory T cells was shown.138 The nonmitochondrial pathways of death contributing to memory cell generation may involve death receptors. For instance, CD4+ T-cell help is required for CD8+ T-cell memory. In the absence of CD4+ help, TNF-related apoptosis-inducing ligand (TRAIL) is expressed on and mediates apoptosis of CD8+ cells upon antigen restimulation.139 TRAIL deficiency, however, only delays the loss of CD8+ memory cells, revealing that non-TRAIL mechanisms also contribute to the homeostasis of CD8+ memory cells.140 Further studies are needed to dissect the PCD mechanisms contributing to T memory cell generation and homeostasis.
B-Cell Homeostasis
We have focused most of our attention on T-lymphocyte apoptosis thus far because it has received the greatest experimental examination and more details are known. However, PCD also governs B-cell homeostasis and is regulated in ways that have similarities and differences with T cells. Both DR triggering and withdrawal of trophic stimuli contribute to B-cell elimination. Similar to T cells, B cells developing in the bone marrow undergo a series of proliferative expansion and apoptotic contraction events to shape the final B-cell repertoire.141 Immunoglobulin (Ig) gene rearrangement starting at the pro-B-cell stage generates the BCR. The cytokine IL-7, which promotes the survival of developing thymocytes,142,143 is also crucial for sustaining survival at the pro-B/pre-B juncture.144,145 IL-7 induces transcription of the antiapoptotic molecule Mcl-1, which selectively binds to and antagonizes the proapoptotic Bcl-2 family member Bim.146,147 Then selection for B cells occurs in two developmental steps with successive gene rearrangements at the heavy and light chain Ig loci. B cells that fail to generate a productive BCR, due to abortive Ig gene rearrangements, are eliminated by PCD. At the other end of the spectrum, B cells that express BCRs directed against self-antigens are eliminated by apoptosis or undergo receptor editing to acquire new antigen specificity.148 The survival and antigen responsiveness of immature B cells can also be increased by lipopolysaccharide exposure, presumably to enhance B-cell production during infections.149 Fas, TNFR-1, and perforin are not involved in the death of developing B cells.150 Rather, death appears to be a consequence of a direct signaling event generated by the BCR.151
In contrast to B cells in development, mature B cells rely heavily on TNFR family receptors such as CD40, Fas, and BAFF for regulating their survival and death. It is also
clear that T cells control PCD of mature B cells. Christopher Goodnow and colleagues have illustrated the importance of CD40 and Fas in the balance of life and death in B cells using the hen egg lysozyme (HEL)-transgenic and HEL-specific TCR transgenic mice.152,153 Naïve, HEL-specific B cells undergo proliferation in the presence of HEL and HEL-specific CD4+ T-cell help. Hence, proper B-cell activation requires triggering of the BCR as well as “help” in the form of CD40L expressed on activated T cells. The primary function of CD40L is to prevent anergy and/or death of activated B cells in the germinal center, allowing further differentiation and function.154 However, anergic HEL-specific B cells from HEL transgenic animals undergo apoptosis in the presence of the same HEL-specific CD4+ T-cell help. This antigen-specific B cell death is absent in Fas-deficient B cells,152 thus suggesting a role for Fas in causing the death of anergic antigen-specific B cells. Fas killing of mature B cells can also be abrogated by BCR engagement and IL-4, which promote antibody responses.155,156 An imbalance of CD40 and Fas signals might contribute to autoantibody production consequent to Fas and FasL mutations in both human and mouse. Indeed, ablation of Fas in murine B cells alone recapitulates the same autoimmune and lymphoproliferative phenotypes noted in lpr/gld mice, illustrating the importance of Fas in enforcing B-cell tolerance.157
clear that T cells control PCD of mature B cells. Christopher Goodnow and colleagues have illustrated the importance of CD40 and Fas in the balance of life and death in B cells using the hen egg lysozyme (HEL)-transgenic and HEL-specific TCR transgenic mice.152,153 Naïve, HEL-specific B cells undergo proliferation in the presence of HEL and HEL-specific CD4+ T-cell help. Hence, proper B-cell activation requires triggering of the BCR as well as “help” in the form of CD40L expressed on activated T cells. The primary function of CD40L is to prevent anergy and/or death of activated B cells in the germinal center, allowing further differentiation and function.154 However, anergic HEL-specific B cells from HEL transgenic animals undergo apoptosis in the presence of the same HEL-specific CD4+ T-cell help. This antigen-specific B cell death is absent in Fas-deficient B cells,152 thus suggesting a role for Fas in causing the death of anergic antigen-specific B cells. Fas killing of mature B cells can also be abrogated by BCR engagement and IL-4, which promote antibody responses.155,156 An imbalance of CD40 and Fas signals might contribute to autoantibody production consequent to Fas and FasL mutations in both human and mouse. Indeed, ablation of Fas in murine B cells alone recapitulates the same autoimmune and lymphoproliferative phenotypes noted in lpr/gld mice, illustrating the importance of Fas in enforcing B-cell tolerance.157
More recent studies using similar transgenic systems suggest that whereas Fas can mediate apoptosis of activated B cells by engaging CD4+ T cells, BCR-induced death of immature or resting mature B cells is primarily Fas-independent. As in T cells, differential expression of pro- and antiapoptotic Bcl-2 family members control mature B-cell fate at different stages. For example, Bcl-2 and Bcl-xL are critical for naïve B-cell survival, whereas Mcl-1 sustains germinal center B cells that persist into memory B cells.158 As for T cells, knockout mouse experiments reveal that both Bim and Puma are central regulators of mature B cell PCD.159,160,161,162 The critical B-cell survival and maturation factor BAFF, in combination with BCR signaling, ultimately controls mature B-cell homeostasis via NF-&kgr;B-mediated induction of antiapoptotic genes like A1 and Bcl-xL.163 BAFF signaling also promotes naïve B-cell survival by sustaining the MAPK signals that block Bim accumulation.164
The attenuation of B-cell responses at the end of an immune reaction is likely to involve cytokine withdrawal death similar to that of T cells, but this phenomenon is less well characterized. Deprivation of cytokines such as IL-7 and IL-15, which enhance cellular survival through the upregulation of Bcl-2, Bcl-XL, or Mcl-1, may be responsible.147,165 Lymphokine withdrawal death in B cells, like T cells, requires de novo ribonucleic acid/protein synthesis.166 An interesting observation is that a secreted lipocalin, identified through microarray analysis of an IL-3-dependent pro-B-cell line, was implicated as a potential mediator for IL-3 withdrawal death.167 This protein appears to regulate intracellular iron levels, which may modulate Bim levels for apoptosis induction.168 However, it remains to be seen whether lipocalins or other similar molecules are involved in lymphokine withdrawal death in primary mature B or T cells. As we will discuss later in this chapter, survival genes such as Bcl-2 can protect cells against lymphokine withdrawal death and therefore play an important role in mature B-cell homeostasis. Moreover, Bcl-2 can contribute to the survival of B lymphoma cells as revealed by the t(14:18) translocation of Bcl-2 to the Ig locus in follicular B-cell lymphomas.169
Dendritic Cell Homeostasis
Understanding the cell fate regulation of DCs is still in its infancy. However, emerging evidence suggests that these highly efficient antigen-presenting cells are also subject to homeostatic regulation by PCD. The natural turnover of DCs was demonstrated in the mouse by Jenkins and coworkers.170 Using elegant cell-labeling experiments, they showed that antigen-laden DCs stimulate the formation of a cluster of activated antigen-specific T cells and then disappear. This process was antigen- and T cell-dependent. Later, it was shown that the TNF homologue TRAIL, which is produced by activated T cells, could induce apoptosis in DCs and that this mechanism could be defective in patients harboring mutations in caspase-10.171 Together, these data introduced the concept that there is homeostatic regulation of DCs involving differentiation and recruitment followed by their active elimination by stimulated T cells. Early removal of DCs has the benefit of allowing the activation of T cells but avoiding T-cell restimulation and propriocidal death too soon in the response to antigen. Recent work in mice genetically engineered to selectively express the p35 apoptosis inhibitory protein in DCs reveals that apoptotic removal of DCs is essential to preserve tolerance and prevent autoimmunity.172 Moreover, forced p35 expression in DCs accelerated autoimmunity on the autoimmune-prone MRL background, similar to what is observed for T- and B-cell apoptosis defects. Thus, impaired DC apoptosis can collaborate with other genetic and environmental factors to powerfully predispose to autoimmunity. Similar to B cells, specific ablation of Fas in DCs also gives rise to autoimmunity and excessive lymphocyte proliferation in mice.157
On the other hand, PCD of DCs may also serve to promote inflammation and clearance of intracellular pathogens. This is achieved through a distinct pathway of PCD known as “pyroptosis.”173 This pathway is selectively induced in DCs and macrophages upon caspase-1 activation in inflammasomes, cytosolic protein complexes designed to detect microbial components and other “danger” signals. Although it shares some morphologic features with apoptosis, pyroptosis is a distinct proinflammatory, lytic process designed to expose intracellular pathogens and recruit neutrophils. Thus pyroptosis appears necessary for effective clearance of intracellular pathogens that would otherwise replicate and avoid immunosurveillance.
Impaired Programmed Cell Death in Lymphoproliferative Disease and Autoimmunity
Genetically modified mice continue to provide invaluable insights into the molecular underpinnings of PCD in immune cells. The recent description of Bim-deficient lpr
mice provides overwhelming evidence that Fas and Bim are the most important proapoptotic genes governing immune homeostasis.174,175,176 Loss of either Fas or Bim function in isolation manifests in lymphoproliferative disease and autoimmunity. Loss of both results in strikingly accelerated fatal lymphocyte accumulation, early-onset lupus-like autoimmune disease, and failed contraction of antiviral T-cell responses. These findings indicate that Fas and Bim must cooperate in ensuring immune homeostasis through proper PCD. This corroborates earlier work showing that lpr combined with Bcl-2 overexpression caused a synergistic increase in lymphadenopathy and autoimmunity.57,58
mice provides overwhelming evidence that Fas and Bim are the most important proapoptotic genes governing immune homeostasis.174,175,176 Loss of either Fas or Bim function in isolation manifests in lymphoproliferative disease and autoimmunity. Loss of both results in strikingly accelerated fatal lymphocyte accumulation, early-onset lupus-like autoimmune disease, and failed contraction of antiviral T-cell responses. These findings indicate that Fas and Bim must cooperate in ensuring immune homeostasis through proper PCD. This corroborates earlier work showing that lpr combined with Bcl-2 overexpression caused a synergistic increase in lymphadenopathy and autoimmunity.57,58
The study of inherited human lymphoproliferative diseases has further cemented the importance of FAS- and BIM-mediated PCD in maintaining immune homeostasis. Autoimmune lymphoproliferative syndrome (ALPS) was the first genetic disorder of impaired lymphocyte apoptosis (and perhaps the first inherited disease of apoptosis ever described) initially linked to debilitating mutations in FAS. We now appreciate the existence of several forms of ALPS caused by germline or somatic mutations in FAS, FASL, and caspase-10.177 A similar syndrome known as RAS-associated autoimmune leukoproliferative disease is caused by somatic gain-of-function mutations in NRAS, which impair lymphokine withdrawal apoptosis by suppressing upregulation of BIM.102 Patients with ALPS typically present early in life with lymphadenopathy and splenomegaly, including the unique expansion of atypical CD4- CD8- “doublenegative” T cells. Autoimmune cytopenias and increased incidence of lymphoma are also characteristic of ALPS and RAS-associated autoimmune leukoproliferative disease, underscoring the significance of Fas- and Bim-induced apoptosis for culling excess lymphocytes to maintain tolerance and prevent malignancy.
Other lymphoproliferative disorders with ALPS-like features have been linked to faulty PCD. The recently recognized immunodeficiency linked to loss of caspase-8 (caspase-8 deficiency syndrome) also features abnormal lymphocytosis due to impaired Fas-induced apoptosis. The unexpected requirement for caspase-8 in lymphocyte activation explains why patients with caspase-8 deficiency syndrome suffer from recurrent infections rather than autoimmune manifestations.178 Similarly, lymphoproliferation and increased susceptibility to bacterial and viral infections characterizes with a recently-identified family with homozygous FADD-deficiency, reflecting a critical function for FADD in Fas-independent immune cell effector functions.179 Excessive accumulation of T cells deficient in signaling lymphocytic activation molecule-associated protein in X-linked lymphoproliferative disease is also ultimately the result of impaired RICD, explained by insufficient induction of FASL and BIM.81 Defective RICD may partially explain why many patients with X-linked lymphoproliferative disease succumb to fulminant infectious mononucleosis upon Epstein-Barr virus infection, marked by lethal overexpansion of activated, Epstein-Barr virus-specific CD8+ T cells that fail to die after repeated encounters with Epstein-Barr virus-infected B cells. Furthermore, defective PCD may be partially responsible for T-cell hyperproliferation and multiorgan infiltration in patients with immunodysregulation, polyendocrinopathy, and enteropathy, X-linked syndrome, and the equivalent “scurfy” mouse model. In this disease, Treg cells are lost due to Foxp3 mutations, hampering polyclonal, Bim-directed T-cell apoptosis induced by Treg-mediated consumption of IL-2.116 As new pathologic mutations are rapidly uncovered by rapid whole-genome sequencing, similar disorders featuring unwanted accumulation of activated lymphocytes, including self-reactive autoimmune clones, may eventually be tied to related defects in cell death programs. The investigation of these disorders of apoptosis and immune homeostasis has provided useful lessons on contemporary human genetic investigation which have been summarized elsewhere.180
Programmed Cell Death and Development of Lymphoid Malignancy
Here we return to the question of why the mature immune system assiduously eliminates activated lymphocytes that have proven their utility in recognizing and responding to specific antigen. Why can’t the immune system adopt a laissez-faire economy of previously activated cells, which might have great value if the same pathogen is re-encountered? Several lines of evidence strongly suggest that in addition to the potential loss of tolerance and autoimmunity, lymphoid malignancy is also promoted by defective apoptosis. Hence, the accumulation of immune cells that have a propensity to proliferate and can undergo additional genetic changes may be deleterious to the organism. The association of translocations of the Bcl-2 gene with diffuse large B cell lymphoma suggested that somatic aberrations in apoptosis pathways might be important in the transformation process. Since then, somatic changes in a variety of apoptosis molecules including Fas, caspases, and Bcl-2 family members have been documented in lymphoid tumors.181 By contrast, the well-known inheritable apoptosis defect in p53 in the Li-Fraumeni syndrome seems to cause predominantly solid tumors but not lymphomas. Examination of large kindreds of patients with ALPS has revealed a 15-fold greater incidence of lymphomas relative to the general population. It is striking that these families have several different classes of lymphoma, suggesting that Fas protects against transformation not for a single cell type but generally for B- and T-lymphocytes.182 Moreover, Fas stimulation not only results in death induction, but also stimulation of signals for survival and proliferation such as NF-&kgr;B and MAPK activation; these dichotomous signals may have different thresholds that can contribute to lymphomagenesis.183 Patients with X-linked lymphoproliferative disease are also at greater risk for developing B-cell lymphomas, which may be linked to impaired B-cell PCD attributed to poor T or natural killer cell cytotoxic capability, including hampered FasL induction.184 Thus, besides autoimmune manifestations, we can infer that protection against lymphoid malignancy demands strict control over the accumulation of activated lymphocytes.
Programmed Cell Death as an Immune Effector Mechanism
Although this topic will be covered authoritatively elsewhere in the book, it is important to recognize that the same pathways that participate in homeostatic cell death also are used as immune effector mechanisms. The Fas pathway is the principal calcium-independent pathway of cytotoxic T-lymphocyte killing.68 Fas ligand displayed by either CD4+ or CD8+ T cells can eliminate Fas-bearing cells that may be infected or malignant. Similarly, the calciumdependent cytotoxic T-lymphocyte killing mechanism involving perforin and granzymes also can induce PCD in target cells. The two main proteases are granzyme A and granzyme B, the latter being the only serine protease capable of processing caspases.185,186,187,188,189 Following endocytosis into a target cell, perforin forms pores in endosomal membranes through which granzyme B gains access to the cytoplasm and proteolytically activates caspases, leading to apoptosis.190 Though this cytoplasmic pathway of death does not emanate from the mitochondria, the latter may amplify the death signal by Bid cleavage.191,192,193,194,195 These parallels may provide an insight into why lymphocytes have evolved mechanisms in trans, involving the surface interaction of FasL and Fas or perforin/granzyme release to homeostatically control their numbers by apoptosis. This permits the T cell to simultaneously regulate itself and other immune cells, and to expunge nonlymphoid cells that require immune elimination such as those that are infected or malignant. Natural killer cells may also use PCD mechanisms for selfregulation and expunging infected or malignant cells.196,197 Hence, for mature T cells, the same molecules can subserve several death functions. By contrast, a more direct connection of the TCR or BCR to death pathways is present in developing lymphocytes, which have no use for effector mechanisms or propriocidal regulation.
Programmed Cell Death as Immune Therapy
Because of its exquisite antigen specificity, the possibility of using antigen-induced death, particularly of T-lymphocytes, for the treatment of immunologic diseases has been suggested.198,199 In particular, the T-cell components of graft rejection, autoimmune diseases, and allergic reactions could potentially be suppressed by antigen-induced elimination. This concept has been tested in both mice and monkeys with clearly beneficial effects on disease outcome. For example, it was found that the repetitive administration of a myelin basic protein antigen could suppress experimental allergic encephalomyelitis in mice by deleting the diseasecausing T cells.93 This suggests that antigen-specific therapy is achievable if antigens relevant to the disease process are sufficiently well-defined and death of the culpable T cells can be triggered without exacerbation of symptoms.29
Because these requirements have limited further evaluation of antigen-specific therapy in human clinical trials, other ways of triggering death have been explored. Fas or TNFR1 agonist administration results in systemic toxicities limiting their potential utility.200,201 However, TRAIL receptor agonists potentially induce death of neoplastic cells preferentially.202,203 Intracellular apoptosis signaling molecules targeted in preclinical or clinical trials include caspases, IAPs, SMAC, and proapoptotic or antiapoptotic Bcl-2 family members.204,205,206,207 Many of these are rationally designed small molecules or peptidomimetics, which have demonstrated promising results, particularly against lymphoid and other malignancies.
CELLULAR AND MOLECULAR MECHANISMS OF PROGRAMMED CELL DEATH
Apoptosis Initiation Mediated by Caspase Complexes: Two Principal Pathways
Caspases must be highly active within the cytoplasm to cause apoptosis. Like all proteases, these potentially destructive proteins are first produced as zymogens that are then proteolytically activated. Elucidating these complex mammalian pathways provides a window into the myriad of molecular abnormalities of apoptosis that contribute to immunologic diseases and cancer (Fig. 30.3). Also, it is necessary to understand the important nonapoptotic functions assigned to caspases including cellular activation, suppression of necrotic pathways, and cytokine processing as key enzymatic functions of inflammasomes. Fortunately, mammalian apoptosis can be understood with a few key concepts. Most importantly, the processing enzymes that activate caspases are mainly caspases themselves. As explained in the following, activation and autoprocessing occur when caspase zymogens are brought into specific signaling complexes. In general, there are two main forms of caspases. Those with long prodomains that have protein interaction domains capable of bringing them into activating platforms for autoprocessing are called initiators, upstream, or apical caspases. Those with short prodomains that must be cleaved by other proteases (caspases or granzyme B) are called effector, downstream, or executioner caspases. Key adaptor molecules harbor domains that specifically recruit caspases into complexes: the death effector domains (DEDs) or the caspase recruitment domains (CARDs).208,209 The DEDs and CARDs are protein-protein interaction domains found in the caspase prodomains and other adapter molecules that generate the specific intermolecular assemblies. In contrast to executioner caspases, which require processing to become active, initiator caspases are activated by multimerization.210,




Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
