Chapter Outline
Severe Combined Immunodeficiency
Other Forms of Combined Immunodeficiency with Variable Presentation
MANAGEMENT OF COMBINED IMMUNODEFICIENCY
OTHER IMMUNODEFICIENCIES ASSOCIATED WITH DEFECTIVE MECHANISMS OF DNA REPAIR
IMMUNODEFICIENCIES WITH CHROMOSOMAL INSTABILITY
MUTATIONS OF THE NUCLEAR FACTOR κ-B PATHWAY
DEFECTS OF TOLL-LIKE RECEPTOR SIGNALING
THE SYNDROME OF WARTS, HYPOGAMMAGLOBULINEMIA, INFECTIONS, AND MYELOKATHEXIS
HYPERIMMUNOGLOBULIN E SYNDROME CAUSED BY STAT3 MUTATIONS
AUTOSOMAL RECESSIVE HYPER-IMMUNOGLOBULIN E SYNDROME CAUSED BY TYK2 MUTATIONS
CHRONIC MUCOCUTANEOUS CANDIDIASIS
IMMUNODEFICIENCIES WITH SKELETAL ABNORMALITIES
IMMUNODEFICIENCIES ASSOCIATED WITH DISORDERS OF FOLATE AND COBALAMIN METABOLISM
IMMUNODEFICIENCIES WITH IMMUNE DYSREGULATION
AUTOIMMUNE POLYENDOCRINOPATHY-CANDIDIASIS-ECTODERMAL DYSTROPHY SYNDROME
IMMUNE DYSREGULATION POLYENDOCRINOPATHY ENTEROPATHY X-LINKED SYNDROME
DEFECTS OF IL-2–MEDIATED SIGNALING
AUTOIMMUNE LYMPHOPROLIFERATIVE SYNDROME
IMMUNODEFICIENCIES WITH IMPAIRED CELL-MEDIATED CYTOTOXICITY
FAMILIAL HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS
X-LINKED LYMPHOPROLIFERATIVE DISEASE
IL-2 INDUCIBLE TYROSINE KINASE DEFICIENCY
IMMUNODEFICIENCIES WITH SELECTIVE PREDISPOSITION TO PATHOGENS
Deficiency of Classical Complement Pathway Components
Deficiency of Alternative Complement Pathway Components
Deficiency of Lectin Pathway Complement Components
Deficiency of Terminal Complement Components
Deficiency of Complement Regulatory Factors
Immune compromise arises either as a heritable genetic defect (primary immune deficiency diseases, PIDDs) or as a consequence of another pathologic process such as infection, malignancy, malnourishment, or iatrogenic immunosuppression (secondary immune deficiencies). Immune dysfunction in patients with PIDDs is most often manifested as recurrent and chronic infections; however, malignancy and autoimmunity are common and may be prominent in some disorders. In many cases, symptoms and signs of immunodeficiency develop soon after birth or early in childhood. However, in some forms of PIDD clinical onset may be delayed into late childhood, adolescence, or adulthood. Even though many patients with severe immunodeficiency suffer dramatic morbidity and mortality in infancy and early childhood, the majority of patients with immunodeficiency have milder forms that permit survival into adulthood, but often with reduced longevity because of infection, autoimmune disease, malignancy, or constitutional debility. Health-related quality of life is significantly impaired in individuals with PIDDs.
PIDDs may be classified with respect to the specific immune effector mechanisms disrupted. In broadest terms, one may distinguish: (a) disorders that affect both cellular and humoral components of specific immunity (combined immunodeficiencies); (b) defects of antibody production (antibody deficiency syndromes): (c) disorders of innate immunity (phagocytic cell defects, disorders of Toll-like receptor (TLR) signaling, complement deficiencies). Moreover, some forms of PIDD are characterized by prominent immune dysregulation. Finally, in some cases impairment of immune function is part of a broader spectrum of symptoms (syndromic immunodeficiency). Discussed here are a broad range of PIDDs arising from aberrant development and function of T and B cells, disorders with immune dysregulation, immunodeficiency syndromes, and complement deficiencies. Phagocytic disorders are the subject of Chapter 22 . Table 24-1 lists several Internet sites with information relevant to primary immunodeficiencies.
| URL ( http:// +) | Name/Description |
|---|---|
| bioinf.uta.fi/idr/Immunology.shtml | ImmunoDeficiency Resource, University of Tampere, Finland |
| www.aaaai.org | American Academy of Allergy, Asthma, and Immunology |
| www.esid.org | European Society for Immunodeficiencies |
| www.immunodeficiencysearch.com | Searchable database, clinical algorithms, laboratory resources |
| www.info4pi.org | Primary Immunodeficiency Resource Center (sponsored by the Jeffrey Modell Foundation) |
| www.ipidnet.org | Immune Phenotyping in Primary Immunodeficiency |
| www.ipopi.org | International Patient Organization for Primary Immunodeficiencies |
| www.jmfworld.org | Jeffrey Modell Foundation (connects directly to Primary Immunodeficiency Resource Center) |
| www.primaryimmune.org | Immune Deficiency Foundation |
| www.usidnet.org | US Immunodeficiency Network (USIDNET) |
Combined Immunodeficiencies
Severe Combined Immunodeficiency
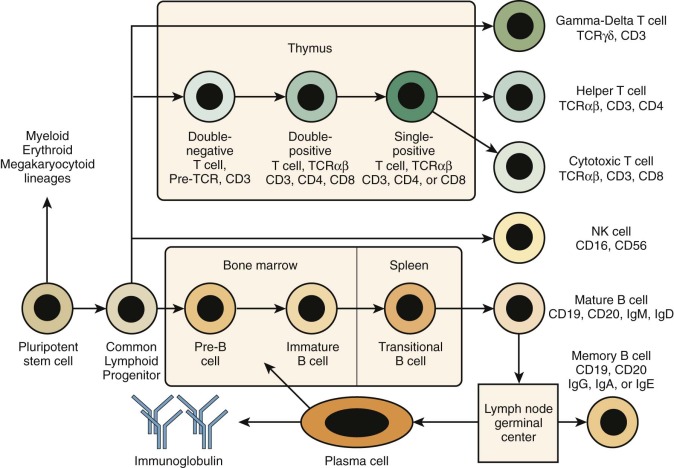
The term severe combined immunodeficiency (SCID) designates a genetically heterogeneous group of PIDDs characterized by impaired development and function of T lymphocytes. In some cases, development of B or natural killer (NK) lymphocytes (or both) is also affected ( Fig. 24-1 ). However, because T cells play a critical role also in most B-cell responses, serious T-cell dysfunction precludes effective humoral immunity even in patients in whom the genetic defect does not affect B-cell development. Complete absence of specific cellular and humoral immunity in patients with SCID leads to an extreme infectious diathesis early in life. Mucocutaneous candidiasis is a common finding. Infections with common viral pathogens (e.g., varicella, herpes simplex, measles, adenoviruses, respiratory syncytial virus, influenza, and parainfluenza) are often fatal. Patients are also susceptible to opportunistic infections with commensal organisms that are normally nonpathogenic, such as Pneumocystis jiroveci. Even attenuated vaccine organisms such as oral polio vaccine virus, rotavirus vaccine, varicella vaccine, and bacille Calmette-Guérin (BCG) can cause severe or fatal infection. Transfusion of blood products containing viable lymphocytes may lead to fatal graft-versus-host disease. Autoimmunity and other manifestations of immune dysregulation (e.g., skin rash) are frequently observed, especially in patients with residual development of T lymphocytes (“leaky” SCID). Patients with SCID are also at increased risk of malignancies, including lymphoproliferative disorders caused by Epstein-Barr virus (EBV) and other tumors.
Typical symptoms of SCID are recurrent severe infections, chronic diarrhea, and failure to thrive. Symptoms are often seen in the first weeks of life but may be delayed by several months. Antibodies derived from the mother by transfer across the placenta may provide some early protection. Physical examination may reveal foci of infection (e.g., thrush) and the absence of discernible lymphoid tissue. Hypogammaglobulinemia is often present but is not universal. Specific antibody responses are almost always severely impaired.
SCID is most often classified according to the peripheral blood lymphocyte phenotype—that is, absence or presence of the major lymphocyte types. T cells are absent (or very low) and nonfunctional in all classic forms of SCID, a phenotype designated “T − .” The presence or absence of B cells is indicated by adding “B + ” or “B − ,” respectively. The same pattern applies for NK cells. Table 24-2 lists the various lymphocyte phenotypes of SCID and the molecular defects associated with them. In as many as 40% of patients with SCID, maternal T cells may have engrafted in the fetus during gestation, and this situation may occasionally confuse the diagnostic picture. Maternally derived T cells tend to be anergic, but they may also be associated with clinical manifestations similar to those of graft-versus-host disease, such as eczematous rash, eosinophilia, and splenomegaly. In rare circumstances, maternal engraftment may lead to even more unusual clinical findings, such as IgA monoclonal gammopathy, autoimmune cytopenias, and allograft rejection.
| Lymphocyte Phenotype | Genes |
|---|---|
| T − B − NK − | ADA, AK2, NP * |
| T − B − NK + | DCLRE1C , LIG4, NHEJ1 , PRKDC, RAG1 , RAG2 |
| T − B + NK − | IL2RG , JAK3 |
| T − B + NK + | CD3D, CD3E, CD3G, † CD3Z, IL7R, PTPRC |
* B-cell development is variably affected in nucleoside phosphorylase (NP) deficiency.
† CD3G mutations are rarely associated with a severe T-cell deficiency.
T − B − NK − Severe Combined Immunodeficiency
Adenosine Deaminase and Purine Nucleoside Phosphorylase Deficiencies.
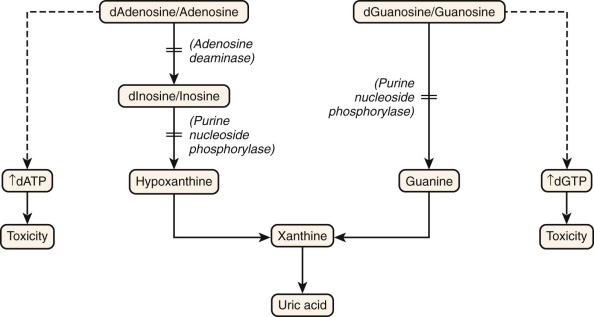
Figure 24-2 shows the salvage pathway of purine nucleotide synthesis. Deficiencies of the enzymes adenosine deaminase (ADA) and purine nucleoside phosphorylase (PNP) are associated with immunodeficiency. ADA deficiency accounts for approximately 10% to 15% of all patients with SCID, or approximately a third of SCID cases with autosomal recessive inheritance. PNP deficiency is quite rare, with approximately 70 cases reported. Although PNP deficiency is mainly expressed as a somewhat selective defect in cellular immunity, it is discussed here because of similarities in their biochemical pathophysiology with ADA deficiency. The absence of ADA or PNP leads to intracellular accumulation of deoxyadenosine and deoxyguanosine, respectively. These molecules are not themselves toxic to lymphocytes. However, when they are converted to their 5′-triphosphates (dATP and dGTP), they inhibit ribonucleotide reductase and prevent de novo synthesis of deoxynucleotides. Without building blocks for the replication and repair of DNA, the cells cease to divide. Lymphocytes die to a much greater extent than other cell types do. In both ADA and PNP deficiency, T-cell precursors in the thymus appear to be especially sensitive to death by apoptosis (programmed cell death). B cells are more often depleted in ADA deficiency than in PNP deficiency. In the latter the B-cell phenotype is more variable.
Approximately 85% of individuals with ADA deficiency will display a SCID phenotype with markedly reduced numbers of both T and B cells and low serum antibody levels. NK cells are only rarely found in patients with ADA deficiency. Additional clinical manifestations may include radiographic alterations of the ribs, vertebral bodies, and iliac crests as well as sensorineural deafness, liver dysfunction, lung disease resembling alveolar proteinosis, and cognitive impairment and other neurologic abnormalities. Immune dysregulation is common in patients with ADA deficiency ; it may manifest with allergy (eczema, asthma) and autoimmunity (cytopenias, hypothyroidism, diabetes) and is associated with loss of regulatory T-cell function. Rarely, ADA deficiency may present with generalized erythroderma and infiltration of skin by activated autologous T cells, resembling Omenn syndrome (OS). Bone marrow hypocellularity is frequently observed in ADA deficiency; myelodysplasia also has been reported. Finally, patients with ADA deficiency are at increased risk for multicentric dermatofibrosarcoma protuberans, a very rare tumor with a characteristic chromosomal translocation (t[17;22][q22;q13]) resulting in the COL1A1 -platelet-derived growth factor β (PDGFB) fusion gene. Between 10% and 15% of patients with ADA deficiency may have a delayed or late-onset form that may be manifested in late infancy or early childhood. These patients initially may have variable numbers of circulating lymphocytes and some humoral immunity that quickly wanes. The severity of the phenotype appears to correlate to some degree with the amount of residual ADA activity. Autoimmunity is particularly common in patients with residual ADA enzymatic activity.
Patients with PNP deficiency are very susceptible to viral and fungal infections. They have decreased numbers of circulating T cells, often initially with normal numbers of B cells and normal serum immunoglobulin (Ig) levels. With time, humoral immunity usually deteriorates. Approximately 50% of children with PNP deficiency have neurologic complications such as spasticity, diplegia, paresis, and other motor disorders, as well as cognitive impairment. Autoimmune manifestations, especially cytopenias, are also very common.
After clinical suspicion is aroused, diagnosis of ADA or PNP deficiency is not difficult. Both ADA and PNP activity is readily measurable in red blood cell or leukocyte lysates. In symptomatic patients, activity is usually 1% or less of that in normal subjects. These methods may be applied also for prenatal diagnosis on cultured amniotic or chorionic villus cells or on fetal blood sampling. PNP is required for the production of hypoxanthine, a precursor of uric acid (see Fig. 24-2 ). Thus individuals with PNP deficiency will have reduced serum and urine urate levels.
Several therapeutic options are available to patients with ADA deficiency, including hematopoietic stem cell transplantation (HSCT), enzyme replacement therapy (ERT) and gene therapy. Excellent results (survival >85%) have been reported after HSCT from matched sibling donors, but results of HSCT from haploidentical or unrelated donors are much less satisfactory (43% and 66% survival, respectively). Moreover, patients with ADA deficiency surviving after HSCT remain at high risk of neurologic complications. ERT with weekly intramuscular injections of polyethylene glycol–conjugated bovine ADA (PEG-ADA) is usually well tolerated and is effective to normalize levels of toxic phosphorylated (deoxy)adenosine derivatives and to improve immune function. However, immune reconstitution is often incomplete. Encouraging results have been obtained with gene therapy, associated with nonmyeloablative cytoreduction. This represents a very important option for patients who lack a human leukocyte antigen (HLA)-identical related donor. HSCT is the only curative therapeutic option for patients with PNP deficiency.
Reticular Dysgenesis Caused by Adenylate Kinase 2 Deficiency.
Reticular dysgenesis caused by adenylate kinase 2 (AK2) deficiency is a very rare autosomal recessive form of SCID, associated with agranulocytosis (with a block at the promyelocyte stage of differentiation in the bone marrow) and sensorineural deafness. AK2 regulates levels of ADP in mitochondria. Deficiency of this enzyme is associated with abnormalities of energy metabolism in granulocytes and lymphocytes (especially T cells), resulting in apoptosis. The number of circulating B and NK lymphocytes can be variably affected, and NK cell differentiation is usually preserved. Therefore, these patients most often exhibit a phenotype other than T- B- NK- SCID; however, we describe this disorder here because it also affects purine metabolism, similar to ADA and PNP deficiencies. OS with oligoclonal expansion of T cells has been described in a patient with AK2 hypomorphic (partially functional) mutations. Patients are at increased risk of myelodysplasia. Sensorineural deafness reflects a role of AK2 in the stria vascularis of the inner ear. Treatment is based on HSCT, but results are less satisfactory than in other forms of SCID, and use of myeloablative conditioning is required for engraftment.
T − B − NK + Severe Combined Immunodeficiency
Defects of Recombinase-Activating Genes (RAG) 1 and 2.
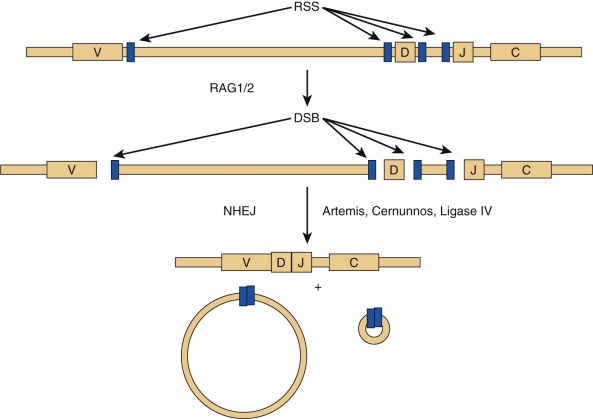
RAG1 and RAG2 encode for DNA-binding proteins that bind to specific recognition sequences flanking coding gene elements of the Ig and T cell receptor (TCR) loci. RAG1 and RAG2 initiate the VDJ recombination process by inducing DNA double-strand breaks at these loci. Proteins of the nonhomologous end-joining (NHEJ) complex ultimately join coding Ig and TCR elements, allowing expression of Ig and TCR molecules ( Fig. 24-3 ). In patients with RAG defects, B- and T-cell differentiation is arrested at an early stage of development. Therefore patients with functionally null mutations of the RAG1 and RAG2 genes have very low numbers of T and B lymphocytes, but NK cell differentiation is preserved. Some patients with hypomorphic RAG1 or RAG2 mutations may sustain differentiation of a limited number of T and (less frequently) B lymphocytes, a condition that is also referred to as “leaky” or “atypical” SCID. In these patients, T lymphocytes often undergo homeostatic proliferation in the periphery and acquire an activated (CD45RO+) phenotype. Oligoclonal expansion of T lymphocytes that infiltrate target tissues causing damage is observed in patients with OS. Patients with OS present with clinical features suggestive of acute graft-versus-host disease. Symptoms include recurrent severe infections, failure to thrive, chronic severe diarrhea, and erythroderma with exfoliation and exudation. Examination may reveal hepatosplenomegaly and lymphadenopathy. Laboratory studies show anemia, hypogammaglobulinemia with an elevated IgE level, a decrease in the number of peripheral B cells, and presence of activated (CD45RO+) circulating T lymphocytes with a restricted (oligoclonal) T cell repertoire. In vitro proliferation of T lymphocytes to antigens and to anti-CD3 is abrogated, whereas proliferation to phytohemagglutinin (PHA) may be variably affected. Although hypomorphic RAG1 and RAG2 mutations are the most common form of OS, the same phenotype may be observed in patients with hypomorphic mutations in other SCID-causing genes. Some evidence suggests that expansion of oligoclonal, tissue-infiltrating T lymphocytes in patients with OS may reflect defects of T-cell tolerance. The study of thymic biopsies from patients with OS has shown abnormalities of medullary epithelial cells, with impaired expression of AIRE (autoimmune regulator, discussed later), a transcription factor that promotes expression of self-antigens, thereby permitting intrathymic deletion of autoreactive T cells or their diversion to FOXP3 + regulatory T (Treg) cells. Consistent with this, absence of FOXP3 + cells has also been demonstrated in thymic biopsies from patients with OS.
Environmental triggers may modify the disease phenotype in patients with RAG gene defects. In at least one case, progression to OS was observed in a patient with a RAG2 mutation after infection with parainfluenza virus type 3. Cytomegalovirus (CMV) infection has been associated with in vivo expansion of TCRγδ + T cells and autoimmune manifestations. Finally, somatic mutations may become superimposed on null or “amorphic” mutations and yield mosaicism with revertant or hypomorphic populations of lymphocytes and resultant OS.
Hypomorphic RAG mutations that result in production of mutated proteins with more robust (although subnormal) recombination activity have been identified in patients with delayed presentation characterized by granulomatous lesions and autoimmunity. Occasionally, autoimmunity in patients with RAG defects is associated with preserved number of circulating B cells and normal or increased Ig serum levels. One patient with compound heterozygous missense RAG1 mutations presented with idiopathic CD4 lymphopenia.
Mutations of DCLRE1C (Artemis).
A form of autosomal recessive SCID with the classic T − B − NK + phenotype has an associated feature of general radiation sensitivity not seen with RAG1 or RAG2 mutations and is caused by mutations in the gene DCLRE1C (DNA cross-link repair enzyme 1C), whose protein product is called Artemis. Artemis has a critical role in the function of NHEJ, or the repair of double-stranded DNA breaks (see Fig. 24-3 ). During VDJ recombination, it opens the hairpins sealing the coding ends at V, D, and J elements that have been targeted by the RAG proteins. Absence or impaired function of Artemis blocks T and B lymphocyte development at an early stage, similar to RAG1 and RAG2 mutations. Artemis is ubiquitously expressed, and loss of its function in nonhemopoietic tissues may account for nonimmunologic problems (oral and genital ulcers, dental abnormalities, and malabsorption) and for the increased cellular radiation sensitivity that these patients exhibit. OS has been observed in some patients with Artemis defects. A partial form of the disease, with residual expression and function of the protein, is characterized by reduced number of circulating T and B lymphocytes and increased risk of lymphoma.
DNA Ligase IV Deficiency.
DNA ligase IV (LIG4) is also required for NHEJ. Mutations in the LIG4 gene are associated with microcephaly, cognitive impairment, bone marrow failure, and cellular immunodeficiency that may manifest as T − B − NK + SCID, “leaky” SCID, or OS. There is profound cellular radiosensitivity. Patients are at increased risk of myelodysplasia and leukemia.
Cernunnos Deficiency.
Cernunnos (also known as XLF) is another component of the NHEJ pathway and is encoded by the NHEJ1 (nonhomologous end-joining 1) gene. Mutations of this gene cause lymphopenia, radiation sensitivity, growth retardation, microcephaly and developmental delay, bone marrow failure, and an increased risk of myelodysplasia. The T-and B-cell immunodeficiency of Cernunnos deficiency is less severe than in patients with RAG or LIG4 defects; however, there are quantitative and qualitative alterations of the T-cell repertoire, a severe reduction in the number of invariant NK T cells and mucosa-associated invariant T lymphocytes, and abnormalities of class-switch recombination in B lymphocytes (see section on class-switch defects).
DNA Protein Kinase Catalytic Subunit (DNA-PKcs) Deficiency.
A homozygous missense mutation in the PRKDC (protein kinase, DNA-activated, catalytic polypeptide) gene, encoding for DNA-PKcs was identified in a patient with T − B − NK + SCID and cellular radiosensitivity, but without microcephaly or mental retardation.
T − B + NK − Severe Combined Immunodeficiency
X-Linked Severe Combined Immunodeficiency, Mutations of the Cytokine Receptor Common γ Chain.
In Western countries, approximately 40% of all patients with SCID are males with an X-linked form of the disease, (XSCID), caused by mutations of the IL2RG (interleukin-2 receptor γ) gene, that encodes for the common γ chain (γ c ), shared by cytokine receptors for interleukin-2 (IL-2), IL-4, IL-7, IL-9, IL-15, and IL-21 ( Fig. 24-4 ). Thus a defect in a single molecule interrupts several cytokine pathways, thereby severely disrupting immune function. Typically, patients with XSCID lack T and NK lymphocytes; these defects reflect defective signaling via IL-7R and IL-15R, respectively. However, some patients with XSCID may have variable numbers of their own T or NK cells and may retain partial immune function, reflecting some preservation of γ c expression and of cytokine-mediated signaling. B lymphocytes are typically present, but they are nonfunctional as a result of impaired IL-21–mediated signaling. Patients with γ c deficiency have typical clinical features of SCID. In addition, they are unusually susceptible to chronic and severe human papillomavirus (HPV) infections, and this phenotype may not be corrected after HSCT. Delayed presentations have been reported in association with hypomorphic mutations or with somatic mutations in T-cell progenitor cells, allowing generation of a diversified pool of functional T lymphocytes that may persist for years. Gene revertant T cells undergoing in vivo expansion may also cause a phenotype resembling OS.
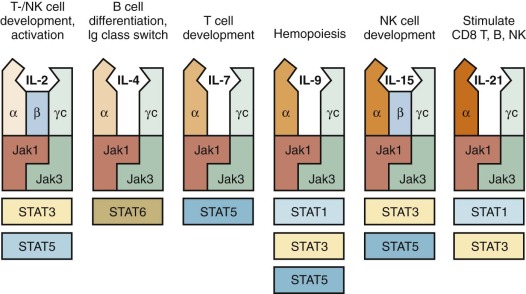
JAK3 Deficiency.
A form of T − B + NK + SCID phenotypically very similar to XSCID is associated with defects in the tyrosine kinase JAK3. The similarity with XSCID is easily understood because JAK3 is a critical signal-transducing molecule associated with the γ c chain (see Fig. 24-4 ). Hypomorphic mutations that allow residual JAK3 protein expression and function may associate with partial preservation of circulating T cells and delayed clinical onset, with lymphoproliferation and susceptibility to warts and other viral infections.
T − B + NK + Severe Combined Immunodeficiency
Defects of the Interleukin-7 Receptor α Chain.
Signaling through the IL-7 receptor (IL-7R) is required for proliferation of early T-cell progenitors and survival of T lymphocytes. The IL-7R is composed of an α chain (IL-7Rα), encoded by the IL7R gene, and γ c (see Fig. 24-4 ). Mutations of the IL7R gene in humans cause T − B + NK + SCID. One case of OS caused by a homozygous missense mutation, permissive for IL-7Rα expression, has been reported.
Mutations Affecting Components of the T-Cell Antigen Receptor Complex
The TCR/CD3 complex is made up of the TCRα and TCRβ (or TCRγ and TCRδ) chains of the receptor making contact with major histocompatibility antigen-peptide complexes, as well as components of the CD3 signal-transducing complex: CD3γ, CD3δ, CD3ε, and CD3ζ ( Fig. 24-5 ).
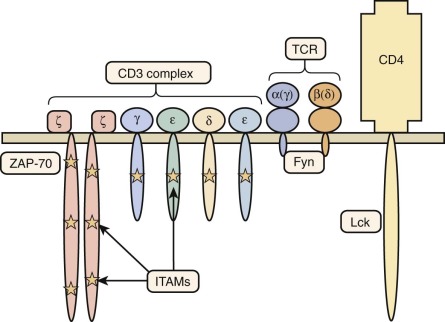
Defects in the CD3δ (gene CD3D ) lead to a T − B + NK + SCID phenotype. Patients have varying degrees of panhypogammaglobulinemia and are susceptible to disseminated viral infections. In some cases, a normal-sized thymus was visualized, in marked contrast to almost all other forms of SCID, in which the thymus is absent or extremely hypoplastic. In one case, CD3δ deficiency was associated with OS. A multicenter study of 13 patients with CD3δ deficiency treated by HSCT has shown superior immune reconstitution after transplantation from HLA-matched donors: use of conditioning chemotherapy may be needed to achieve engraftment.
Deficiency of CD3ε (gene CD3E ) and of CD3ζ (gene CD3Z ) are associated with a similar phenotype and markedly reduced levels of the TCR/CD3 complex on the surface of circulating T lymphocytes. In one patient with CD3ζ deficiency, some of the circulating T cells were found to express normal levels of the TCR/CD3 complex as the result of somatic mutations on one of the CD3Z alleles, allowing expression of poorly functioning TCR/CD3 complexes.
In contrast, the phenotype of CD3γ (gene CD3G ) deficiency is heterogeneous. One of two genotypically affected siblings presented with clinical features of SCID and autoimmunity associated with severe T-cell lymphopenia, whereas the other one had a milder immunologic phenotype and remained asymptomatic until 12 years of age, when he developed autoimmunity and recurrent infections. This variability of clinical and immunologic phenotype has been confirmed in other patients.
A homozygous splice-site mutation in the TCRα constant region gene has been identified in two patients with a history of recurrent infections and autoimmunity. Development of T cells expressing TCRγδ was not affected. In vitro proliferative responses to mitogens and antigens were decreased, but specific antibody production was preserved.
CD45 Deficiency
A few patients have been found to have a form of SCID resulting from mutations in the gene encoding the protein tyrosine phosphatase CD45 (gene PTPRC ), which plays an important role in T-cell activation. These patients have a SCID phenotype with diminished numbers of poorly functional T cells, normal or increased numbers of B cells, and variable number of NK cells.
Other Combined Immunodeficiencies (with T Cells Present)
Table 24-3 lists disorders of combined immunodeficiency with variable lymphocyte phenotypes.
| Syndrome | Gene |
|---|---|
| Variable T cell lymphopenia | CORO1A, MST1 |
| Relative CD4+ T cell deficiency | LCK, MHCIITA , RFX5 , RFXANK , RFXAP, RHOH |
| Relative CD8+ T cell deficiency | CD8A, TAP1 , TAP2 , TAPBP , ZAP70 |
| Hyper IgE syndrome type 2 | DOCK8 |
| Veno-occlusive disease with immunodeficiency | SP110 |
| Ion (Ca ++ and Mg ++ ) transport defects | MAGT1, ORAI1, STIM1 |
| Other | CARD11, IL21R |
Relative Deficiency of CD4+ T Cells
Lck Deficiency.
The lymphocyte-specific protein tyrosine kinase (Lck) associates with CD4 and CD8 molecules and plays a critical role in T-cell signaling by mediating phosphorylation of immunoreceptor tyrosine activation motifs in the intracytoplasmic domains of CD3 subunits and of the ζ chain–associated protein of 70 kDa (ZAP70). Homozygosity for a missense LCK mutation was identified in a child with recurrent respiratory infections, protracted diarrhea, failure to thrive, skin nodules, vasculitis, arthritis, and autoimmune thrombocytopenia. There was a severe defect in the number of circulating CD4+ lymphocytes, and the density of both CD4 and CD8 molecules on the surface of CD3+ cells was markedly reduced. T lymphocytes showed a restricted TCR repertoire. Proliferative responses to anti-CD3 were profoundly impaired.
RhoH Deficiency.
The Ras homology family member H (RhoH) is a small GTPase that is phosphorylated in response to TCR stimulation, allowing recruitment of Lck and ZAP-70 to the TCR-associated signaling complex. Mutations of the RHOH gene have been identified in two siblings with warts, variably associated with granulomatous lung disease, psoriasiform skin changes, and molluscum contagiosum. One of the affected individuals developed Burkitt lymphoma in childhood. Immunologic abnormalities included reduced number of naïve CD4+ lymphocytes and oligoclonal T cell repertoire, with accumulation of CD8+ effector memory CD45RA+ lymphocytes (TEMRA cells) with an “exhausted” (CD45RA+CCR7−) phenotype. The reduced number of lymphocytes expressing skin-homing receptors (cutaneous lymphocyte antigen, CCR4, CCR6, CCR10, β7-integrin) may impair defense against cutaneous viral infections and contribute to development of warts.
Major Histocompatibility Complex Class II Deficiency.
The major histocompatibility complex (MHC) in humans is called the HLA complex. The class I and class II HLA molecules expressed on cell surfaces are critical for practically all mechanisms of specific immune activation. Class I molecules are present on most cells of the body. They present antigenic peptides derived principally from intracellular sources to the antigen receptors of T cells expressing CD8. HLA class II molecules are generally restricted to specialized antigen-presenting cells (APCs), including monocytes and macrophages, dendritic cells, and B cells, but are also expressed by thymic epithelial cells. HLA class II molecules mainly present peptides imported from outside the cell to antigen receptors of T cells bearing CD4. Defects in the synthesis, expression, or function of HLA molecules may be expected to have profound consequences on lymphocyte development and on immune competence. In particular, defective expression of HLA class II molecules on the surface of thymic epithelial cells impairs development of CD4+ lymphocytes by impairing their positive selection.
Most patients with MHC class II deficiency present early in life with recurrent and severe infections, chronic diarrhea, and failure to thrive. Chronic opportunistic infections of the gastrointestinal tract caused by Cryptosporidium or CMV organisms may lead to severe liver disease with sclerosing cholangitis. The number of circulating CD4+ cells is markedly decreased, and delayed hypersensitivity responses are impaired. All forms of MHC class II molecules (DP, DQ, and DR) are not expressed. Most patients have profound hypogammaglobulinemia; however, some patients have normal serum Ig levels, yet specific antibody responses are not elicited. As a rule, these children do not survive without HSCT. However, bone marrow transplantation in these patients has not been consistently successful, probably reflecting inability to correct defective expression of MHC class II molecules on the surface of thymic epithelial cells.
MHC class II deficiency results from defective regulation of gene transcription as a result of mutations in any of four different genes (CIITA, RFXANK, RFX5, RFXAP) encoding for components of HLA class II transcription complex. Without a complete transcription complex, no HLA class II mRNA is produced, and class II proteins cannot be synthesized. The same complex activates transcription at all HLA class II gene promoters. RFXANK mutations are the main cause of the disease, which is more common in Northern Africa and in the Middle East.
Relative Deficiency of CD8+ T Cells
Defective Expression of HLA Class I.
Several patients with defective expression of HLA class I molecules have been described. These patients have mutations in one of two genes encoding either TAP1 or TAP2 (transporter associated with antigen processing 1 or 2) or in the gene encoding tapasin, a TAP-binding protein molecular chaperone. The TAP1/TAP2 heterodimer is required for shuttling of cytosolic peptides across the endoplasmic reticulum to load onto nascent HLA class I molecules for eventual expression at the cell surface and presentation to TCRs ( Fig. 24-6 ). HLA class I molecules are highly unstable without a peptide in their binding cleft, and they are not transported to the cell surface. Patients with defective expression of HLA class I molecules suffer mainly from upper and lower respiratory tract bacterial infections, bronchiectasis, and granulomatous skin inflammation. Occasional asymptomatic individuals have also been described. The main immunologic abnormality is low peripheral blood CD8+ T cells; antibody responses may be normal.
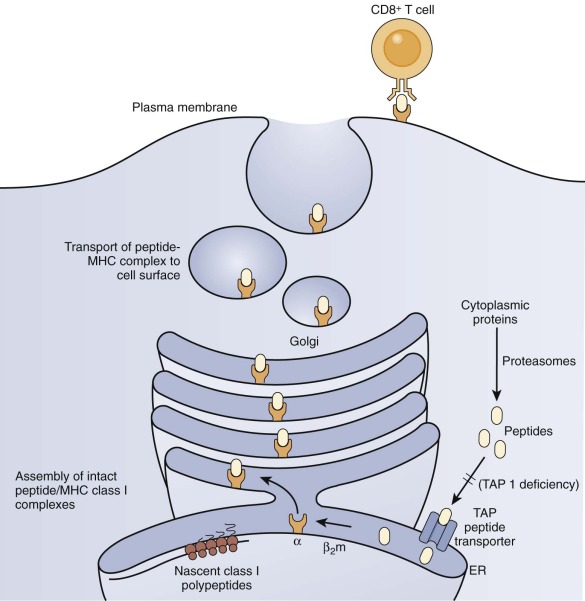
ZAP-70 Deficiency.
The ZAP-70 tyrosine kinase has a critical role in TCR-mediated signaling by inducing phosphorylation of the linker of activated T cells and SH2 domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76) (see Fig. 24-5 ). Mutations of the ZAP70 gene in humans are typically associated with severe, early onset infections. Patients have almost complete absence of CD8+ T cells. Although circulating CD4+ cells appear normal, they fail to respond to signals delivered by way of the TCR. In a few cases, hypomorphic ZAP70 mutations have been associated with delayed onset of the disease and manifestations of immune dysregulation (skin rash, wheezing, eosinophilia). Treatment is based on HSCT.
CD8α Deficiency.
CD8 molecules are expressed on the cell surface as either a CD8α 2 homodimer or a CD8αβ heterodimer. CD8α (gene CD8A ) deficiency is a rare autosomal recessive disorder characterized by lack of circulating CD8+ lymphocytes and by an increased number of CD3+CD4−CD8− cells that express markers of effector cytotoxic T lymphocytes (CD11b, CD57). The disease is reported here because of some phenotypic similarities (reduced number of CD8+ cells) with ZAP-70 deficiency; however, clinical manifestations are distinct and more similar to MHC class I deficiency, with recurrent respiratory infections and delayed diagnosis.
Other Forms of Combined Immunodeficiency with Variable Presentation
A number of primary immunodeficiencies may have a variable clinical phenotype; in some cases, patients affected with these disorders may present with clinical and laboratory features suggestive of a combined immunodeficiency.
Macrophage Stimulating 1 Deficiency
Macrophage stimulating 1 (MST1), also known as serine threonine kinase 4 (STK4), is a molecule that controls apoptosis and survival of naïve T lymphocytes by inducing expression of FOXO1, a transcription factor that induces expression of the IL-7R. MST1 deficiency is associated with recurrent infections, immune dysregulation, a moderate degree of neutropenia, and congenital heart disease. Viral infections are particularly common and include warts, molluscum contagiosum, and EBV-driven lymphoproliferative disease. The number of naïve T cells is markedly decreased, and there is an expansion of TEMRA cells, associated with a restricted T cell repertoire. T lymphocytes show increased apoptosis and reduced proliferation to mitogens. There is also an increased proportion of transitional B lymphocytes, with reduced number of memory B cells.
Dedicator of Cytokinesis 8 Deficiency
Dedicator of cytokinesis 8 (DOCK8) is a guanine nucleotide exchange factor that activates Cdc42 and participates at intracellular signaling in lymphoid and dendritic cells. DOCK8 mutations cause an autosomal recessive form of combined immunodeficiency with hyper-IgE (this is also known as hyper-IgE syndrome type 2). Patients suffer from chronic viral cutaneous infections (warts, molluscum contagiosum), recurrent skin abscesses and cellulitis, respiratory tract infections, candidiasis, eczema and other manifestations of severe allergy, autoimmunity (cytopenias, central nervous system [CNS] vasculitis), and an increased risk of malignancy (epithelial cell carcinoma, lymphomas, leiomyosarcoma). The disease has a severe prognosis with high mortality rate; treatment is based on HSCT.
Immunologic abnormalities include T-cell lymphopenia with impaired thymopoiesis, a low number of naïve CD8+ cells, and accumulation of TEMRA lymphocytes. T cell proliferation to mitogens and antigens is often reduced, especially within CD8+ lymphocytes. Ig abnormalities include elevated serum IgE and low levels of IgM, and B cell responses to TLR agonists are compromised. Immunologic memory is defective in both T- and B-cell compartments. In most cases, the genetic defect is represented by large intragenic deletions.
Coronin-1A Deficiency
The Coronin-1A protein inhibits F-actin formation in T lymphocytes and thereby modulates cytoskeleton rearrangement and T-cell activation. Coronin-1A deficiency causes a combined immunodeficiency with recurrent and severe infections, including severe varicella and EBV-driven lymphoproliferation. Developmental delay has also been observed, possibly reflecting a role for Coronin-1A in the CNS. There is profound T-cell lymphopenia, with reduced survival of T lymphocytes and impaired in vitro proliferation to mitogens and antigens. HSCT can cure the immunologic abnormalities and the increased susceptibility to infections.
Veno-Occlusive Disease with Immunodeficiency Syndrome
Veno-occlusive disease with immunodeficiency (VODI) syndrome is an autosomal recessive condition characterized by severe infections (often including Pneumocystis jiroveci and CMV infection) and hepatomegaly with abnormalities of liver function and evidence of veno-occlusive disease on liver biopsy. Cerebrospinal leukodystrophy has been reported in several patients. Immunologic abnormalities include T- and B-cell lymphopenia, poor T-cell function, severe hypogammaglobulinemia, and lack of memory B lymphocytes. If untreated, the disease is often fatal within the first years of life. Administration of intravenous immunoglobulin (IVIG) and antimicrobial prophylaxis may prolong survival, but the only curative approach is represented by HSCT. Recurrence of veno-occlusive disease after HSCT has been reported. VODI was initially described in patients of Lebanese descent, but the disease has been reported in other ethnic groups as well. The disease is caused by mutations of SP110 encoding for a nuclear body protein that is thought to have a role in regulation of gene transcription.
Immunodeficiencies Caused by Defects of Calcium-Release Activated Channels
On TCR-mediated signaling, Ca ++ is mobilized from endoplasmic reticulum (ER) stores. This depletion is sensed by stromal interacting molecule 1 (STIM1), which oligomerizes and binds to the ORAI proteins that form the calcium-release activated channels (CRACs) on the cell surface, thereby permitting influx of Ca ++ , activation of transcription factors (e.g., nuclear factor of activated T cells), cytokine production, T-cell proliferation, and induction of effector T cell responses. Mutations of STIM1 and ORAI1 genes in humans account for autosomal recessive combined immunodeficiencies with recurrent infections, signs of ectodermal dystrophy, nonprogressive myopathy (reflecting a role of CRACs in muscle cells). Autoimmune manifestations (cytopenias, lymphadenopathy, and splenomegaly) may also be observed, especially in patients with STIM1 deficiency. Kaposi sarcoma due to human herpesvirus 8 (HHV-8) infection has been reported. The absolute lymphocyte count is normal, with preserved distribution of major lymphocyte subsets. However, in vitro proliferation to mitogens and antigens is impaired, and cell-mediated cytotoxicity and cytokine production are also affected.
Magnesium Transporter Protein 1 Deficiency
Magnesium transporter protein 1 (MAGT1) is an X-linked disease characterized by severe chronic viral infections, chronic diarrhea, and EBV-driven lymphoproliferative disease. Immunologic abnormalities include CD4 lymphopenia and impaired T cell proliferation to CD3 stimulation. The MAGT1 gene encodes for a membrane-associated Mg ++ transporter. Female carriers have a skewed pattern of X chromosome inactivation in circulating T lymphocytes, suggesting that Mg ++ signaling plays an important role in T lymphocyte development and survival.
Caspase Recruitment Domain Family Member 11 Deficiency
Caspase recruitment domain family member 11 (CARD11) is a scaffold protein that interacts with B-cell lymphoma 10 and mucosa-associated lymphoid tissue lymphoma translocation gene 1 proteins, thereby inducing activation of the canonical nuclear factor κB (NF-κB) pathway in lymphocytes. Biallelic loss of function mutations of CARD11 are associated with a combined immunodeficiency, marked by increased susceptibility to opportunistic infections and hypogammaglobulinemia. The number and distribution of T lymphocytes are preserved, other than for a reduced number of Treg cells; by contrast, there is expansion of transitional B lymphocytes and reduction of mature naïve and memory B lymphocytes. Moreover, in vitro activation of T and B cells with phorbol myristate acetate is severely impaired, and proliferation of T lymphocytes in response to stimulation by way of CD3/CD28 is abrogated. On in vitro activation, production of IL-2 by T cells and of interferon IFN-γ by NK cells is decreased.
IL-21 Receptor Deficiency
IL-21 regulates proliferation of lymphocytes, differentiation of T helper 17 (Th17) cells, maturation of B cells into plasmablasts, and induction of NK cell cytotoxicity. IL-21 receptor (IL-21R) defects have been identified in four patients from two kindreds whose clinical phenotype included respiratory infections, chronic diarrhea, failure to thrive, and severe liver and biliary tract disease caused by infection with Cryptosporidium organisms. The number of T, B, and NK lymphocytes is normal, but the proportion of switched memory B cells is reduced. Serum IgG levels are low or slightly subnormal, but IgE is elevated. Antibody responses to T-dependent and T-independent antigens are decreased. On in vitro activation with CD40 ligand (CD40L) and IL-21, B lymphocytes from the patients show reduced proliferation and defective class switch recombination. NK cytotoxic activity is markedly decreased. T cells proliferate normally in response to stimulation by CD3 and CD28 but fail to produce IL-17 and IL-22. Proliferation to antigens in vitro is impaired.
Management of Combined Immunodeficiency
Diagnostic Approach
Suspicion of SCID or other forms of combined immunodeficiency is prompted by clinical symptoms, family history, or positive results of screening assays. In particular, newborn screening for SCID can be performed by quantifying levels of TCR excision circles (TRECs; see Fig. 24-3 legend) in dried blood spots collected at birth. TRECs are a by-product of rearrangement of TCR genes during intrathymic T-cell development. In patients with SCID, TREC levels are extremely low or absent as a result of lack of significant thymic T-cell output. Indeed, since newborn screening for SCID has been started in various parts of the United States, several cases of SCID have been identified at birth in otherwise healthy-looking newborns. However, it is not clear yet whether determination of TREC levels at birth may also reliably identify other forms (and which ones) of combined immunodeficiency with residual T-cell development. Tandem mass spectrometry can also be used for newborn screening of ADA deficiency and may also identify subjects with residual levels of ADA activity, who could otherwise be missed by the TREC-based assay.
Quantification of TRECs represents an important, yet nonspecific, indicator of a possible severe T-cell immunodeficiency, the diagnosis of which must be confirmed by appropriate immunophenotypic, functional, and molecular tests. Flow cytometry, looking at the distribution and absolute number of the major T-cell subsets, B and NK lymphocytes, may provide important information. Determination of naïve (CD45RA+CD62L+) and activated/memory (CD45RO+ CD62L−) T lymphocytes should be included in the diagnostic panel to correctly identify patients with maternal T-cell engraftment or with leaky SCID and expansion of in vivo activated, activated T cells. Functional tests include in vitro proliferation to mitogens (PHA, anti-CD3 monoclonal antibody) and antigens (provided that the patient has been exposed to these through immunization or natural infection). Ultimately, identification of the specific disease-causing mutation(s) may permit molecular diagnosis of the disease.
Hematopoietic Stem Cell Transplantation
Identification of a patient with SCID should be considered a pediatric emergency. Children with this disorder are so prone to infections that extreme measures must be taken to protect them from microbial invasion. Before newborn screening for SCID became a reality, most patients were already infected when initially evaluated and required aggressive interventions to keep their clinical course from running inexorably downhill. After full immunologic evaluation has been performed (as expeditiously as possible), these patients should begin receiving IVIG infusions. Prophylaxis for P. jiroveci pneumonia is indicated for all patients.
Although alternative therapies may work for some patients (e.g., PEG-ADA for ADA deficiency), the definitive treatment for the great majority of patients with SCID and other combined immunodeficiencies is HSCT. A variety of sources have been used for stem cells, including HLA-identical sibling donors, haploidentical related donors (usually parents), unrelated donors, and umbilical cord blood. Transplantation from a matched sibling donor in patients with SCID does not require conditioning and is associated with excellent survival and long-term immune reconstitution. Patients with SCID who lack a matched sibling donor may receive HSCT from one of their haploidentical parents, most often the mother. In this case, mature T lymphocytes must be removed from the graft to prevent graft-versus-host disease. T cell–depleted HSCT from a haploidentical donor for SCID may be performed without or with pretransplant chemotherapy. Unconditioned T cell–depleted haploidentical transplantation leads to long-term T-cell engraftment ; however, B- and NK-cell engraftment is rarely achieved, and lifelong use of IVIG may be needed, especially in patients who lack B cells (e.g., defects of VDJ recombination) or those whose genetic defects compromise B-cell function (γc, JAK3 defects). Use of chemotherapy may allow stem cell engraftment (and hence also permit reconstitution of B- and NK-cell immunity); however, it is also associated with toxicity. In a large series, overall survival in SCID patients transplanted after the year 2000 was 90% when HLA-matched siblings served as donors and 66% after haploidentical HSCT. However, when transplantation is performed in infants younger than 3.5 months of age, survival is greater than 95%, even after haploidentical donors, thus justifying newborn screening for SCID based on quantification of TRECs at birth.
HSCT is also the mainstay of treatment for other forms of combined immunodeficiency with presence of residual or normal number of autologous T lymphocytes. In such cases, however, pretransplant conditioning must be used to achieve engraftment. Overall survival after HSCT for these conditions is less satisfactory than in patients with SCID, being approximately 80% from matched siblings and 50% from haploidentical donors.
Gene Therapy
Gene therapy for the treatment of human disease was first attempted in patients with ADA deficiency. A functional copy of the ADA gene in a retroviral vector was transduced into populations of mature T cells or bone marrow cells. Transduced cells expressed normal levels of ADA and showed normal function in vitro. However, clinical efficacy was difficult to assess in these early attempts because adjunctive therapy with PEG-ADA was mandated on ethical grounds, even after the re-introduction of transduced cells into patients. More recently, gene therapy for ADA deficiency has been performed by transducing hematopoietic stem cells without concurrent in vivo administration of PEG-ADA ; use of submyeloablative doses of busulfan has permitted robust engraftment and long-term immune reconstitution in the vast majority of the patients. However, the absolute T-cell count remains suboptimal in many patients, possibly reflecting irreversible thymic damage caused by toxic adenosine derivatives.
The earliest “complete” success of gene therapy is considered to have occurred with XSCID. Hematopoietic stem cells were transduced ex vivo with a retroviral vector carrying a functional copy of the γ c gene and then infused into patients without any conditioning chemotherapy. Twenty patients have been treated in this manner. Most patients have exhibited rapid and persistent reconstitution of T cell–mediated immunity. Reconstitution of B-cell immunity has been achieved only in some patients. The therapy appears to be less effective if performed later in life, probably because of deterioration of the thymic rudiment over time.
Unfortunately, 5 of the 20 infants with XSCID treated by gene therapy have developed leukemia, which was fatal in one. Leukemias were caused by insertional mutagenesis. The vector integrated within or near oncogenes (in particular, LMO2 ) and the strong enhancers contained in the viral long terminal repeats (LTRs) caused oncogene transactivation and uncontrolled cell proliferation. Novel clinical trials, based on self-inactivating retroviral and lentiviral vectors in which expression of the γ c gene is driven by weaker cellular promoters (instead of the strong viral LTRs), are currently under way in Europe and in the United States.
Immunodeficiency Syndromes
The term syndromic immunodeficiencies has been coined to describe the occurrence of abnormal development or function of the immune system leading to susceptibility to infection, autoimmunity, or malignancy in the setting of a genetically determined disorder with characteristic features of altered development or function of other organ systems or the body as a whole. Here we will review several disorders with prominent immune dysfunction. These diseases and their associated genes are listed in Table 24-4 . Space does not permit a detailed description of the tremendous variety of syndromes that have been associated with immunodeficiency. Interested readers are referred to an excellent review on this topic.
| Syndrome | Gene |
|---|---|
| Wiskott-Aldrich syndrome | WAS |
| DiGeorge syndrome | del22q11, TBX1 |
| DNA repair defects: | |
| Ataxia-telangiectasia | ATM |
| Nijmegen breakage syndrome and others | ATLD, NBS1, PMS2 |
| Disorders of chromosome instability: | |
| Bloom syndrome | BLM |
| ICF syndrome | DNMT3B, ZBTB24 |
| Disorders of pigmentary dilution: | |
| Chédiak-Higashi syndrome | LYST |
| Griscelli syndrome type 2 | RAB27A |
| Hermansky-Pudlak syndromes types 2 and 9 | AP3B1, PLDN |
| Defects of the NF-κB pathway | IKBKG , IKBA |
| Defect in Toll-like receptor signaling | IRAK4, MYD88 |
| WHIM syndrome | CXCR4 |
| Hyper IgE syndromes | STAT3, TYK2 |
| Chronic mucocutaneous candidiasis | CARD9, IL17RA, IL17F, STAT1 |
| Immunodeficiencies with skeletal abnormalities: | |
| Cartilage-hair hypoplasia | RMRP |
| Schimke immuno-osseous dysplasia | SMARCAL1 |
| Disorders of vitamin metabolism | TCN2, MTHFD1 |
Wiskott-Aldrich Syndrome
Wiskott-Aldrich syndrome (WAS) is an X-linked disease characterized by eczema, immunodeficiency, and thrombocytopenia. Bloody diarrhea is often seen and may be the initial feature. Eczema may be mild or exuberant; staphylococcal superinfection is common. Recurrent otitis media and sinopulmonary infections occur frequently, and opportunistic infections are also seen. In addition, autoimmune hemolytic anemia, vasculitis, inflammatory bowel disease, glomerulonephritis, and other autoimmune processes have been frequently observed in WAS. Patients most often die as a result of overwhelming infection or massive hemorrhage. Those who avoid these complications have a greatly increased risk for lymphoma, many of which are EBV positive. There is also an increased risk of aortic aneurysms.
Variability of the clinical phenotype has been reported, and a clinical score has been developed to reflect this phenotypic heterogeneity. A milder form of the disease, characterized by thrombocytopenia with mild eczema and lack of severe infections, is also referred to as isolated X-linked thrombocytopenia (XLT). Although XLT patients have better survival rates compared with patients who have classical WAS, nonetheless they have a significant risk of complications (e.g., autoimmunity, hemorrhage). In some cases, the thrombocytopenia may even be intermittent. On the other hand, patients who present early in life with very profound (<10 × 10 9 /L) and persistent thrombocytopenia have very poor outcomes.
Platelets are small, do not function normally, and are also cleared more rapidly (although the number of bone marrow megakaryocytes is normal or even increased). In the appropriate clinical context, measurement of platelet size confirms the diagnosis. In normal individuals platelet volume is 7.1 to 10.5 fL, with a diameter of 2.3 ± 0.12 µm, whereas platelets from patients with WAS have volumes ranging from 3.8 to 5.0 fL with diameters of 1.82 ± 0.12 µm. The low platelet volume distinguishes the thrombocytopenia of WAS from immune thrombocytopenic purpura (ITP), in which platelets are often large. However, ITP may develop in as many as 20% of patients with WAS.
The mainstay of treatment for WAS is HSCT. Without HSCT, survival beyond adolescence is uncommon. The rates for 5-year survival after HSCT for WAS approximates 90% for transplants performed after year 2000. Optimal results (survival >95%) are obtained after HSCT from matched siblings, but significantly improved outcome has been also achieved after HSCT from unrelated donors and even mismatched related donors. Mixed chimerism is associated with increased risk of autoimmunity after transplant.
Pretransplant morbidity and mortality rates may be reduced with regular infusions of IVIG and antibiotic prophylaxis. Blood products, when needed, should be irradiated and tested for CMV. Splenectomy often increases the platelet count, although immune thrombocytopenia may later supervene. Moreover, splenectomy markedly increases the risk of overwhelming infections. Immunosuppressive agents may be needed to treat autoimmune complications of the disease.
The immune defects in WAS are variable. Immune function may be normal in early infancy, but it gradually wanes. T cell numbers may be decreased, and T cells have diminished, but not absent, responses to mitogens and antigens in vitro. T cells in WAS will also proliferate in response to nonspecific activating stimuli such as phorbol esters and calcium ionophores but show a striking absence of reactivity to immobilized antibodies directed against the CD3 complex of the TCR. The diversity of the T-cell repertoire appears normal throughout childhood, but it may become more restricted with consequent worsening immunodeficiency in adulthood.
Serum levels of IgA and IgE are often elevated, whereas IgM may be low; however, increased serum IgM is associated with an increased risk of autoimmunity and worse outcome. Isohemagglutinins and specific antibody responses to polysaccharide antigens are also diminished. Although the frequency of class-switched B cells (i.e., those not expressing IgM) appears to be normal, there are fewer memory B cells expressing CD27, thus indicating some alteration in B-cell activation in germinal centers that could underlie or contribute to the humoral immunodeficiency. Finally, dendritic cells and monocytes show impaired formation of podosomes and defective directional migration in response to chemotactic signals.
The gene defective in WAS has been identified; its official name is WAS, and its product is designated the WAS protein (WASP). The same gene is defective in XLT, as well as in X-linked congenital severe neutropenia. The latter disease is due to activating WAS mutations; its phenotype is very different from WAS and includes chronic neutropenia with a differentiation block in the bone marrow and an increased risk of myelodysplasia.
WASP interacts with several intracellular partners, including the WASP-interacting protein (WIP), Rho family guanosine triphosphatases, and the Arp2/3 complex, which leads to reorganization of the actin cytoskeleton in response to activating stimuli in lymphoid and myeloid cells.
There is a good, yet imperfect, genotype-phenotype correlation in WAS. In particular, XLT is more often associated with residual expression of WASP and with missense mutations in exons 1 and 2 of the WAS gene that correspond to a domain of WASP that interacts with WIP. This interaction stabilizes WASP, thereby explaining reduced levels of WASP detected in cells from patients with XLT. In contrast, the more severe phenotypes are associated with complete absence of protein or with expression of truncated, nonfunctional forms of WASP. Somatic mutations allowing expression of WASP in some cell populations (more often, in CD8+ T lymphocytes) have been frequently reported in WAS. However, the impact of this phenomenon on clinical phenotype remains unclear.
The diagnosis of WAS is facilitated by analysis of WASP expression by flow cytometry or Western-blotting; however, this approach may miss patients who express a mutant form of the protein. Ultimately, mutation analysis is required to confirm the diagnosis. Female carriers of WAS are generally asymptomatic and have normal platelet counts but show nonrandom X-chromosome inactivation in lymphocytes and granulocytes. Occasionally, occurrence of WAS or XLT in females has been reported as the result of various mechanisms, including extreme lyonization favoring the mutant X chromosome, random X-chromosome inactivation, or WAS gene mutation on both X chromosomes.
WAS Protein-interacting Protein Deficiency
A single case of a female patient with mutations of the WIPF1 gene, encoding WIP, has been reported. The phenotype included recurrent infections, thrombocytopenia, defective T-cell proliferation and chemotaxis, and impaired NK cell function. No expression of the WIP and WASP proteins was detected in circulating cells.
DiGeorge Syndrome
DiGeorge syndrome (DGS) arises as a result of a failure of migration of neural crest cells into the third and fourth pharyngeal pouches. Patients display a characteristic facies, cardiac defects, parathyroid hormone deficiency, and varied immune defects. The facies consists of hypertelorism; micrognathia; short philtrum; and low-set, posteriorly rotated ears with small pinnae. Cardiac defects are varied. Type B interrupted aortic arch is the most common and is associated with DGS in 50% of patients with this defect. Truncus arteriosus and other conotruncal anomalies are also common. Structural anomalies of the airways have likewise been found in some patients, as well as dysphagia secondary to laryngoesophageal dysmotility. The bilateral paired parathyroid glands are normally adherent to the thymus. Thus these organs are affected together in this disease. The parathyroid deficiency is often more pronounced than the thymic defect; hypocalcemic tetany is one of the more common initial symptoms of DGS. Even small islands of ectopic thymus tissue may permit the development of sufficient T cells for immune competence. Neuropsychiatric problems and some degree of cognitive impairment are seen in many patients and become more prominent during adulthood.
Partial and complete forms of DGS have been distinguished on the basis of clinical and immunologic characteristics. In the complete form the thymus is absent or severely hypoplastic. These patients have absent or markedly decreased numbers of T lymphocytes. B cells are present, but specific antibody production is impaired. Some patients may have erythroderma, similar to that seen in OS. In this condition, also referred to as atypical complete DGS, oligoclonal, activated (CD45RO+) tissue-infiltrating T lymphocytes are detected. If untreated, complete DGS is most often fatal in early childhood; patients die as a result of hypocalcemia, cardiac complications, infection, or a combination of these causes.
The occurrence of partial forms of DGS outnumbers that of the complete form by about 100 : 1. T-cell number and function in most patients are varied and presumably correlate with the amount of ectopic thymus tissue present. Immune function in patients with complete DGS does not improve; patients with partial DGS may have less immunodeficiency as they get older. In some cases, the diagnosis has been made in adults being evaluated for hypoparathyroidism and hypocalcemia. Humoral immunity is generally intact in patients with partial DGS; however, IgA deficiency and defective antibody production have been reported. An increase in atopic dermatitis and asthma has been reported in individuals with 22q11 deletion. Autoimmunity (especially ITP, other cytopenias, juvenile arthritis, and celiac disease) is observed in 10% of the patients.
In various series, between 55% and 90% of patients with DGS have 2 to 3 megabase deletions involving chromosome 22q11.2. Deletions in the same region are found in patients with a spectrum of phenotypically overlapping conditions, including velocardiofacial syndrome and conotruncal anomaly face syndrome, and in some patients with isolated cardiac defects. There is heterogeneity in expression of the DGS phenotype, even in monozygotic twins. Approximately 40 genes are present in the deleted regions in DGS, and the contributions of each to the phenotype are only beginning to be understood. TBX1 encodes a member of a family (T-box) of transcription factors. Mice that are heterozygous for deletion of this gene have a phenotype that is variable but often very similar to that of humans with DGS, including thymic and parathyroid hypoplasia, cardiac outflow tract abnormalities, and abnormal facial structure. Two truncating and three missense mutations in TBX1 have been found in a few patients with DGS or velocardiofacial syndrome and no detectable 22q11.2 deletion. Interestingly, the missense mutations were found to result in gain of function, suggesting that increased TBX1 activity may phenocopy TBX1 loss of function or haploinsufficiency. Some patients with DGS have deletions on chromosome 10p13-p14.
Although recurrent infections can be observed in up to 25% to 30% of patients with DGS, severe viral and opportunistic infections are very rare, other than in patients with complete DGS. Live viral vaccines may be administered safely to the great majority of patients. By contrast, complete DGS has a severe prognosis, but it can be treated successfully by thymus transplantation. Immunosuppression is needed to control severe immune dysregulation in patients with complete atypical DGS while preparing for thymus transplantation. In a recent study 43 of 60 patients (72%) treated with this procedure were reported to be alive at a median of 4.7 years after transplantation, and they attained an increase in T-cell count with improved function, although absolute naïve CD4+ and CD8+ counts remained below the 10th percentile in most of them. HSCT from matched donors may also provide some immune reconstitution through homeostatic proliferation of mature T lymphocytes contained in the graft; however, in a series of 17 patients with complete DGS treated by HSCT, overall survival rate was only 41% and the count of naïve CD4+ cells remained very low.
FOXN1 Deficiency
The transcription factor FOXN1 plays a critical role in development of the thymus and eccrine glands. FOXN1 mutations in humans cause athymia, profound T-cell lymphopenia, alopecia totalis, and nail dystrophy, fully resembling the “nude” phenotype observed in Foxn1 -mutated mice. There is complete lack of CD4+ lymphocytes, whereas a low number of oligoclonal CD8+ cells may be present. Similar to DGS, reconstitution of T-cell immunity can be achieved with thymic transplantation. Alternatively, HSCT from HLA-identical siblings may provide some immune reconstitution through homeostatic proliferation of T lymphocytes contained in the graft.
Ataxia-Telangiectasia
Ataxia-telangiectasia (AT) is an autosomal recessive disorder with an estimated incidence of 1 in 20,000 U.S. Caucasian births (95% confidence interval, 1 in 2500 to 700,000). AT is characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, and immunodeficiency. Associated features are an increased incidence of lymphoma and sensitivity to radiation. Impairment in motor development may be noted early in the disease course and is progressive. Walking may be delayed until 16 to 18 months of age or later. Telangiectases of conjunctival and cutaneous vessels do not appear until children are 3 to 5 years of age. The cutaneous lesions are found mainly on the pinnae and in skin creases. Immunodeficiency will occur in approximately 70% of patients. Manifestation of the immunodeficiency is quite varied, but it is often seen as recurrent sinopulmonary bacterial infections. Granulomatous lesions are frequently seen and may reflect immune dysregulation. Growth retardation is a prominent feature in many patients and has been associated with decreases in insulin-like growth factor I and its binding protein.
The median age at death in patients with AT is approximately 19 to 25 years. Many patients die as a result of progressive pulmonary disease caused by repeated infection. Possibly, noninfectious and ultimately fatal interstitial lung disease may also occur in approximately 25% of patients. The lifetime risk for malignancies in AT is 30% to 40%; 85% of the cancers are acute leukemias or lymphomas. Elevated serum levels of α-fetoprotein (AFP) are found in more than 95% of patients with AT and have diagnostic value in the appropriate clinical context; however, levels of AFP are physiologically higher at birth and for the first two years.
T and B cell lymphopenia is a well-recognized, though variable, feature of AT in adulthood but may also be present at birth and be detected by abnormal TREC levels at newborn screening for SCID. T cells bearing the TCRγδ constitute up to 50% or more of the total number of T cells in patients with AT. These cells make up fewer than 10% of peripheral T cells in normal individuals. Cutaneous delayed hypersensitivity responses, as well as results of in vitro assays of T-cell function, are often depressed. Varying degrees of humoral immunodeficiency are common. IgA deficiency occurs in two thirds of patients. Decreased levels of IgG2, IgG4, and IgE are also common, yet approximately 10% to 30% of the patients have increased IgM. Impaired specific antibody responses, particularly to pneumococcal polysaccharide antigens, have also been noted in AT patients.
The gene that is defective in AT is designated ATM (AT mutated). ATM has critical roles in regulation of the cell cycle: It is involved in the detection of DNA damage and in the initiation of repair. There is a marked delay in induction of the tumor suppressors p53 and BRCA1 after exposure of AT cells to radiation. Even low-dose radiation leads to significantly greater DNA damage in AT fibroblasts and lymphoblasts than in normal individuals. ATM mutations are associated with impaired lymphocyte proliferation. Lymphocytes of patients with AT also show a remarkable number of chromosomal translocations and inversions. These changes predominantly involve the loci that rearrange to generate mature Ig (2p12, 22q12, and 14q32) and TCR (7p15, 7q35, and 14q11) genes and reflect defective repair of the double-stranded breaks produced as part of the physiologic gene rearrangement process during lymphocyte development. ATM is also involved in class-switch recombination (CSR), and this may explain reduced levels of IgG, IgA, and IgE seen in patients.
No definitive cure is available for AT. Medical management aimed at preventing and treating infections (antibiotics, Ig), tumor surveillance, and neurorehabilitation represent the mainstays of treatment. Very recently, some improvement of neurologic signs has been observed on administration of betamethasone, possibly reflecting antiinflammatory and antioxidant activity of glucocorticoids in the CNS.
Whether pathologic changes result from the heterozygous carrier state of AT remains a subject of controversy. Carriers heterozygous for ATM mutations have none of the classic clinical manifestations of AT; however, the sensitivity of their DNA to radiation is increased, and several reports suggest that AT heterozygotes have an increased rate of a variety of malignancies, such as breast cancer.
Other Immunodeficiencies Associated with Defective Mechanisms of DNA Repair
The Nijmegen breakage syndrome is a disorder of chromosome fragility with autosomal recessive inheritance. The phenotype is similar to that of AT and consists of growth retardation, microcephaly, immunodeficiency, sensitivity to radiation, and a high rate of lymphoma. The affected gene is designated NBS1 . Nibrin, its protein product, is another substrate of ATM in the cellular response to ionizing radiation. Absence of nibrin function leads to disruption of mechanisms of DNA repair, but it does not appear to result in abnormal function of cell cycle checkpoints. Another similar syndrome, AT-like disorder, results from mutations of the gene MRE11A , which encodes another ATM substrate.
Postmeiotic segregation 2 (PMS2) and MutS homologue 6 (MSH6) are components of the DNA mismatch repair pathway. PMS2 deficiency is associated with increased occurrence of malignancies, café-au-lait spots, and defective CSR. MSH6 deficiency is also associated with increased risk of cancer and defective maturation of antibody responses, with impairment of CSR and a skewed pattern (higher rate of transitions) of somatic hypermutation.
Immunodeficiencies with Chromosomal Instability
Bloom syndrome is characterized by chromosomal instability with increased sister chromatid exchange and homologous chromosome translocations and is associated with increased frequency of infections, low levels of IgM, and impaired delayed-type hypersensitivity responses. The disease is caused by mutations of the BLM gene, encoding for a helicase.
The immunodeficiency-centromeric instability facial anomalies syndrome is characterized by hypogammaglobulinemia, facial dysmorphisms, and branching of chromosomes 1, 9, and 16. The disease may be caused by mutations in the DNA methyl transferase 3B (DNMT3B) or in the zinc-finger- and-BTB (bric-a-brac, tramtrack, broad complex)-domain-containing 24 (ZBTB24) genes.
Mutations of the mini-chromosome maintenance-deficient 4 (MCM4) gene have been identified in patients with recurrent and severe viral infections (in particular, caused by herpes simplex virus 1 [HSV-1], varicella zoster virus [VZV] and CMV), growth retardation, adrenal insufficiency, and selective NK cell deficiency with lack of the CD56 dim NK subset. The disease is inherited as autosomal recessive trait. MCM4 is a component of a multiprotein complex required for both initiation and elongation of eukaryotic DNA replication. MCM4 mutations were shown to cause abnormalities of cell cycle, with increased proportion of fibroblasts in G2/M and S phases of cell cycle and an aberrant (>4n) DNA content, and genomic instability.
Pigmentary Dilution Disorders
Pigmentary dilution disorders include diseases in which abnormalities in melanosome trafficking within cells lead to characteristic pale skin and silvery-gray or ashen hair. Several forms of pigmentary dilution disorders are also characterized by a variable degree of immune deficiency, in particular impaired cell-mediated cytotoxicity. In these conditions inability to clear virus-infected cells leads to persistent and exaggerated inflammatory responses with macrophage activation, release of proinflammatory cytokines, and continuous activation of infiltrating CD8 + T cells that secrete high amounts of IFN-γ and cause tissue damage. Fever, hepatosplenomegaly, lymphadenopathy, and cytopenia caused by impaired hematopoiesis in the bone marrow and hemophagocytosis are typical manifestations of immune activation during the so-called accelerated phase of the disease. Impaired cytotoxicity of virus-infected cells reflects defective trafficking, docking or release of cytolytic granules in NK cells, and cytotoxic T cells. This defect parallels defects of melanosome trafficking.
Chédiak-Higashi syndrome is characterized by partial albinism, peripheral neuropathy, and increased risk of viral infections. Neutropenia is often present and may facilitate bacterial infections. Recognition of “giant” lysosomes in leukocytes facilitates the diagnosis. The disease is caused by mutation of the lysosomal trafficking regulator (LYST) gene. The precise mechanism by which the LYST protein works is not known, although it is thought to regulate trafficking of intracellular vesicles. HSCT is the only definitive treatment, but it does not prevent possible progression of neurologic complications.
Griscelli syndrome (GS) type 2 also associates partial albinism and immunodeficiency with increased susceptibility to infections, fever, and cytopenia. The disease is caused by mutations of the RAB27A gene, which encodes for a protein that permits docking of the cytolytic granules to the cell membrane. In contrast, GS type 1 (caused by mutations of the MYO5A gene, encoding for the myosin Va protein) and GS type 3 (due to mutations of the gene encoding for the melanin chaperone melanophilin) are characterized by hypopigmentation without immunodeficiency.
Hermansky-Pudlak syndrome type 2 (HPS2) is characterized by oculocutaneous albinism, bleeding diathesis, neutropenia, interstitial lung disease, and recurrent infections. Patients are at increased risk of hemophagocytosis. The disease is caused by mutations of the β subunit of the adaptor-related protein complex 3 (gene AP3B1 ) involved in sorting of proteins to the lysosomes. Patients with HPS9, which is caused by mutations of the pallidin (PLDN) gene, present with hypopigmentation and nystagmus; recurrent cutaneous infections also have been reported.
Mutations of the Nuclear Factor κ-B Pathway
NF-κB is a transcription factor that is critical for the activation of numerous genes on leukocyte stimulation. Activity of this factor is regulated by an inhibitor known as IκB. Phosphorylation of IκB by IκB kinase (IKK) leads to IκB degradation and liberates active NF-κB. IKK is composed of three subunits, α, β, and γ; the latter is also known as the NF-κB essential modulator (NEMO). The gene encoding NEMO is on the X chromosome. Null mutations in this gene are incompatible with life for male subjects and lead to the X-linked dominant disease incontinentia pigmenti in females. Certain mutations that allow partial NEMO function are associated with an immunologic phenotype that may have some similarities to hyper-IgM. Along with a combined immunodeficiency, these patients may exhibit hypohidrotic ectodermal dysplasia and lymphedema with osteopetrosis. Distinct hypomorphic mutations define specific disease characteristics, thus establishing genotype-phenotype correlation, albeit imperfectly. Somatic mosaicism in T lymphocytes has been detected in a significant proportion of males with NEMO deficiency, providing evidence for selective advantage for NEMO-expressing T cells in vivo. However, whether this may affect the disease phenotype is not clear.
Patients may suffer opportunistic-type disseminated viral infections and P. jiroveci pneumonia and are also highly susceptible to atypical mycobacterial infections. Inflammatory bowel disease is also frequently seen, reflecting a role of NF-κB in controlling epithelial integrity and the interaction between the mucosal immune system and gut microflora. Finally, lymphedema and osteopetrosis may also be part of the disease phenotype.
Most patients are hypogammaglobulinemic and have impaired vaccine responses (especially to polysaccharide antigens). Peripheral blood lymphocyte subsets and in vitro measures of lymphocyte function may be unremarkable or variably diminished; NK cell cytotoxicity may be impaired. Signaling by way of TLRs is also affected. Therapy with gamma globulin and antibiotic prophylaxis is essential. Despite these measures, infections may still occur. HSCT may cure the immunologic abnormalities and resolve increased susceptibility to infections, but inflammatory bowel disease may persist, along with extra-immune manifestations of the disease.
A few patients have been found to have a similar phenotype as a result of a so-called hypermorphic (gain of function) mutation of the IκBα chain. In this situation the IκBα is resistant to phosphorylation by IKK and cannot be dissociated, degraded, and induced to release NF-κB so that it may translocate to the nucleus to activate transcription.
Defects of Toll-Like Receptor Signaling
IRAK-4 and Myd88 are components of a complex mediating signaling downstream of all TLRs (except TLR3). Patients with IRAK4 or MYD88 mutations have severe, invasive bacterial infections such as meningitis, bacteremia, septic arthritis, deep-tissue abscesses, and osteomyelitis with gram-positive organisms (predominantly Staphylococcus aureus and Staphylococcus pneumoniae . Less severe skin and respiratory infections also occur frequently. Most patients begin to exhibit infections in early childhood, but these wane significantly in frequency and intensity in the second decade of life. Most patients have impaired antibody responses to pneumococcal polysaccharides, but other routine tests of immune function are normal. Many patients have high levels of all Ig isotypes, possibly owing to immune stimulation by repeated bacterial infections. Signaling is impaired through all TLRs other than TLR3. Management is mainly with antibiotic prophylaxis and IgG therapy.
The Syndrome of Warts, Hypogammaglobulinemia, Infections, and Myelokathexis
The syndrome of warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM syndrome) is an autosomal dominant disorder of leukocyte trafficking caused by mutations of the chemokine receptor CXCR4. Patients carry heterozygous mutations in the intracellular tail of CXCR4 that prevent interaction with β-arrestin, thus allowing continuous signaling induced by CXCR4 ligand, CXCL12. This signal retains mature neutrophils in the bone marrow (myelokathexis) and alters trafficking of B and T lymphocytes, causing lymphopenia.
Impairment of cellular immunity and defective number of plasmacytoid dendritic cells (which are the major source of IFN-γ) cause warts, and neutropenia and hypogammaglobulinemia account for recurrent bacterial infections that mostly affect the respiratory tract. The number of switched memory B cells is reduced, and there is oligoclonality of the antibody repertoire. Gamma globulin replacement therapy generally reduces the rate of infection. The CXCR4 antagonist AMD3100 (plerixafor) corrects neutropenia and lymphopenia.
Hyperimmunoglobulin E Syndrome Caused by STAT3 Mutations
Hyper-IgE syndrome (HIES, often designated type 1) was initially described in 1972 in patients with severe sinopulmonary bacterial infections, severe skin superinfections with S. aureus on a chronic eczematous eruption (for which the disease is also known as Job syndrome ), and susceptibility to Aspergillus fumigatus pneumonitis with pneumatocele formation. Additional manifestations include asymmetric or coarse facial features, delayed shedding of primary teeth, bone fragility and fractures, scoliosis, and keratoconjunctivitis. Patients show increased susceptibility to nonfilamentous molds, such as P. jiroveci pneumonia and in particular chronic mucocutaneous candidiasis (CMC). There is an increased rate of malignancies, especially non-Hodgkin lymphoma. Cardiovascular anomalies have been described in recent years and include aneurysms, in particular of the carotids, cerebral arteries, and coronary arteries, and may lead to additional complications such as myocardial infarction. Inheritance of this form of HIES is autosomal dominant, but many cases represent sporadic presentations. The disease is caused by heterozygous dominant-negative mutations of the STAT3 gene that encodes for a transcription factor involved in signaling from a multitude of receptors. In particular, STAT3 is required for the development of T h 17 cells, which secrete IL-17 and IL-22, two cytokines that play an important role in defense against candida and in promoting secretion of antimicrobial peptides by bronchial epithelial cells.
The major immunologic findings are elevated serum levels of IgE (3000 to >50,000 IU/mL) and eosinophilia. Along with a deficiency of T h 17 cells, a reduced number of memory (CD27+) B lymphocytes also have been reported. The diagnosis is established on clinical and laboratory grounds; a scoring system has been developed that may be helpful, especially if used in combination with demonstration of lack of T h 17 cells. Management of HIES caused by STAT3 mutations is mainly by skin care and antimicrobial prophylaxis against S. aureus . The role of antifungal prophylaxis is controversial, but it may be helpful in patients with CMC. HIES patients may lack classical signs and symptoms of infection, such as fever. Therefore careful physical examination, medical history, and imaging studies are needed to evaluate, diagnose, and treat any infections. Controversial results have been obtained after HSCT.
Autosomal Recessive Hyper-Immunoglobulin E Syndrome Caused by TYK2 Mutations
Two distinct forms of autosomal recessive HIES are known. Of these, DOCK8 deficiency has been discussed in the preceding section “Other Combined Immunodeficiencies with Variable Presentation.” Two patients with autosomal recessive HIES caused by mutations of TYK2 , a member of the JAK family of tyrosine kinases involved in intracellular signaling, have been reported. The first patient presented with atopy, elevated serum IgE, and susceptibility to infections sustained by S. aureus , BCG, Salmonella organisms, and viruses. The second patient suffered from disseminated BCG infection, neurobrucellosis, and cutaneous herpes zoster but had normal serum IgE and did not develop atopy.
Chronic Mucocutaneous Candidiasis
CMC affecting the nails, skin, and mucosa is the clinical hallmark of three distinct genetic disorders that affect IL-17–dependent immunity. These include complete, autosomal recessive deficiency of IL-17 receptor α chain (IL17RA), partial autosomal dominant IL-17F deficiency, and heterozygous gain-of-function mutations of signal transducer and activator of transcription 1 (STAT1) . The latter represents the most common cause of isolated CMC. Mutations of caspase recruitment domain-containing protein 9 (CARD9) gene, encoding a cytosolic adaptor molecule involved in intracellular signaling in cells recognizing fungi, also affect development of T h 17 cells and cause CMC; however, CARD9-deficient patients are also at increased risk of disseminated Candida infections as well as of other fungal infections. Deficiency of T h 17 cells accounts for CMC also in other forms of primary immunodeficiency, including T-cell deficiencies, HIES, and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED).
Immunodeficiencies with Skeletal Abnormalities
Cartilage-hair hypoplasia (CHH) is an autosomal recessive disease characterized by short-limbed dwarfism with metaphyseal dysplasia, sparse and fair hair, and joint hypermotility. Dyserythropoietic anemia and a variable degree of immunodeficiency are present in 80% of the patients. A minority of subjects also present with Hirschsprung disease. There is a 6% to 10% rate of malignancies, especially lymphoma and basal cell carcinoma. The disease has a higher prevalence among Finnish and Amish populations and is caused by mutations in the RMRP gene that encodes for the untranslated RNA component of the mitochondrial RNA-processing endoribonuclease that is involved in multiple functions, including ribosomal and messenger RNA processing, mitochondrial DNA replication, and negative regulation of target RNA transcripts on interaction with TERT (telomerase reverse transcriptase).
The immunodeficiency of CHH is characterized by increased frequency of infections. Severe and persistent viral infections (EBV, CMV, HSV, and VZV) and fungal infections may occur, in addition to recurrent respiratory infections. Granulomatous lesions affecting the skin and other tissues have been described in several patients. Laboratory tests reveal a variable degree of immune dysfunction that predominantly affects T cells. In rare cases, immunodeficiency is very severe and may resemble SCID, “leaky” SCID with reduced number of CD8+ lymphocytes, or OS. Irrespective of the severity of the clinical phenotype, thymopoiesis is reduced, and there is T-cell lymphopenia. Moreover, T lymphocytes show abnormalities of cell cycle progression and an increased rate of apoptosis. HSCT is an effective form of treatment for patients with a severe clinical phenotype.
Schimke immuno-osseous dysplasia is an autosomal recessive disease characterized by spondyloepiphyseal dysplasia, focal glomerulosclerosis leading to progressive renal failure, and T-cell immunodeficiency. There is T cell lymphopenia, with an increased proportion of activated/memory T cells and of TCRγδ-expressing T lymphocytes. The disease is caused by mutations of the SMARCAL1 gene, which encodes for a chromatin remodeling protein.
Immunodeficiencies Associated with Disorders of Folate and Cobalamin Metabolism
Inborn errors of folate and cobalamin metabolism may associate with varying degrees of immunodeficiency. In particular, genetically determined defects in vitamin B 12 metabolism are associated with megaloblastic anemia, neutropenia, and neurologic symptoms with seizures and developmental delay. Transcobalamin II (gene TCN2 ) deficiency causes neutropenia leading to recurrent bacterial infections. Severe lymphopenia is observed in patients with deficiency of methionine synthase, causing a SCID-like phenotype with severe viral and bacterial infections. Mutations of proton-coupled folate transporter that impede folate absorption through the gastrointestinal mucosa are also associated with extreme lymphopenia and clinical features of SCID. Mutations of the MTHFD1 gene that encodes for a trifunctional protein essential for processing of single-carbon folate derivatives cause SCID with decreased number of T, B, and NK lymphocytes, which is associated with megaloblastic anemia, leukopenia, atypical hemolytic uremic syndrome, and neurologic abnormalities. Parenteral supplementation therapy is beneficial.
GATA2 Deficiency
In 2010 Vinh and coworkers reported on a series of patients with a distinct clinical phenotype characterized by onset in late childhood or adulthood, susceptibility to disseminated nontuberculous mycobacterial disease, recurrent and severe viral infections (especially those caused by papillomavirus and herpesviruses), and fungal infections. There is increased risk of myelodysplasia and malignancies, including acute myeloid leukemia, squamous cell carcinoma, and EBV-positive leiomyosarcoma. Pulmonary alveolar proteinosis and primary lymphedema have been observed in several patients.
Typical laboratory features include profound monocytopenia, severe reduction in the number of B and NK lymphocytes (in particular, of the CD56 bright subset) and dendritic cells and elevated levels of fms-like tyrosine kinase ligand (Flt3L). Depletion of regulatory T cells also has been reported. Examination of the bone marrow reveals hypocellularity, often associated with multilineage dysplasia and cytogenetic abnormalities, including monosomy 7 and trisomy 8. In vitro IL-12 and IFN-γ production in response to stimulation with LPS is markedly reduced, suggesting that disruption of the IL-12/IFN-γ axis may contribute to the increased occurrence of mycobacterial disease.
The disease has been referred to as MonoMAC syndrome (to indicate the monocytopenia and susceptibility to nontuberculous mycobacterial infections) or DCML (dendritic cells, monocytes, B and NK lymphocytes) deficiency and has been shown to be caused by heterozygous, loss-of-function mutations of the GATA2 gene. GATA-2 is a transcription factor that contains two highly conserved zinc finger domains that mediate protein-DNA and protein-protein interactions and is required for stem cell homeostasis, lymphatic vessel development, and the maturation and homeostasis of NK lymphocytes. Disease-causing mutations also include intronic deletions and point mutations in regulatory regions of the gene that affect GATA2 transcript levels. Both autosomal dominant inheritance and sporadic presentation have been reported. If untreated, the disease has a severe prognosis, with death occurring in adulthood as a result of infections or malignancies. Nonmyeloablative HSCT has been successfully performed in several patients, with reconstitution of the number of monocyte, B-cell, and NK-cell populations and reversal of the clinical phenotype.
Immunodeficiencies with Immune Dysregulation
Although autoimmune and inflammatory manifestations may be seen in a large number of primary immunodeficiencies, they represent the main feature in some. The pathophysiology of these disorders may reflect a variety of mechanisms, including impairment of central and peripheral tolerance and chronic immune activation associated with the inability to clear infections. These disorders and their associated genes are listed in Table 24-5 .
| Syndrome | Gene |
|---|---|
| Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) | AIRE |
| Immune dysregulation polyendocrinopathy X-linked (IPEX) | FOXP3 |
| Defects of IL-2 signaling | IL2RA, STAT5B |
| Autoimmune lymphoproliferative syndrome (ALPS) | CASP8, CASP10, FAS, FASLG, KRAS, NRAS |
| Other | ITCH |
Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Syndrome
APECED syndrome represents the prototypic disorder associated with impairment of central T-cell tolerance. APECED is inherited as an autosomal recessive trait and is characterized by autoimmune hypoparathyroidism, Addison disease, and CMC. Patients are also prone to other autoimmune manifestations, such as hepatitis, alopecia, ovarian failure, and hypothyroidism. There is an increased incidence of oral squamous cell carcinoma, and fulminant autoimmune hepatitis. The disease is caused by mutations in the gene AIRE (autoimmune regulator) encoding a transcription factor expressed by medullary thymic epithelial cells (mTECs). AIRE promotes expression of tissue-specific antigens that are presented by mTECs and thymic dendritic cells to nascent T lymphocytes. High-affinity recognition of self peptides leads to deletion of self-reactive T cells. Therefore mutations of the AIRE gene compromise the mechanism of central tolerance and result in APECED, which is associated with production of multiple autoantibody specificities. Interestingly, autoantibody production in patients with APECED is not restricted to AIRE-dependent self-antigens but also includes antibodies to interferon-α and β as well as IL-17 and IL-22. Defective T h 17-dependent immune responses may account for chronic mucocutaneous candidiasis in this disease.
Immune Dysregulation Polyendocrinopathy Enteropathy X-Linked Syndrome
Treg cells maintain peripheral T cell tolerance by suppressing self-reactive T lymphocytes that have escaped central tolerance. Generation of Treg cells is controlled by the transcription factor FOXP3. Mutations of the FOXP3 gene cause immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. IPEX is characterized by severe early-onset autoimmune manifestations that involve the gut (causing intractable diarrhea), pancreas (diabetes), the skin, and other organs. Lymphoproliferation and cytopenias are also very common. Delayed presentations have been also observed. Patients with IPEX require immune suppression, but the only definitive treatment currently available is allogeneic HSCT.
Defects of IL-2–Mediated Signaling
IL-2–mediated signaling is important to maintain immune homeostasis and support the function of Treg lymphocytes. Mutations in the IL2RA gene, which encodes CD25, the α chain of the IL-2R, cause immunodeficiency with IPEX-like immune dysregulation. The clinical phenotype is marked by severe diarrhea, failure to thrive, CMV infection, chronic lung disease, lymphadenopathy, splenomegaly, and autoimmunity (diabetes mellitus, autoimmune hepatitis). Poor in vitro T-cell function that could not be corrected with exogenous IL-2 was demonstrated. The number of FOXP3 + cells was preserved in patients with CD25 deficiency; however, secretion of IL-10, a cytokine with immunosuppressive activity, was impaired, possibly accounting for the autoimmune manifestations of the disease.
Mutations of the signal transduced and activator of transcription 5B (STAT5B) gene, which encodes a transcription factor activated by IL-2 signaling, cause an autosomal recessive form of immunodeficiency with immune dysregulation. Because STAT5B is also activated in response to growth hormone receptor stimulation, patients with STAT5B deficiency also show growth failure with growth hormone insensitivity.
Autoimmune Lymphoproliferative Syndrome
Apoptosis of self-reactive lymphocytes is another important mechanism to prevent autoimmunity in the periphery and may be induced either through the extrinsic, Fas-dependent pathway or through induction of the mitochondrial pathway in response to cell damage and cytokine (e.g., IL-2) starvation. Both pathways converge on caspase 9 and downstream effector caspases 3 and 7, causing apoptosis. Autoimmune lymphoproliferative syndrome (ALPS) is characterized by lymphadenopathy, splenomegaly, and autoimmune cytopenias and an increased risk of developing lymphoma. Infiltrative lymphoproliferative disease may also cause organ-specific complications (glomerulonephritis, hepatitis, encephalitis, uveitis, interstitial pulmonary disease). There is a characteristic expansion of CD3+TCRαβ+CD4−CD8− double-negative T cells (DNTs).
Most patients are heterozygous for a mutation in the FAS gene; in such families the disease is inherited as an autosomal dominant trait with variable penetrance. Somatic mutations in FAS are another common cause of the disease. More rarely, patients may show biallelic mutations.
Other rare causes of ALPS are represented by mutations of genes encoding Fas ligand (FasL), FADD, caspase 8, and caspase 10, all of which are involved in the Fas-mediated signaling pathway leading to apoptosis. The number of DNTs is only marginally elevated in patients with caspase 8 deficiency.
In addition to an increased proportion of DNTs, other biomarkers that may facilitate the diagnosis of ALPS and that are associated especially with FAS mutations include elevated serum levels of vitamin B 12 , soluble FasL, IL-10, and IL-18. Hypergammaglobulinemia is common. Moreover, patients with ALPS caused by FAS mutation frequently have a reduced number of memory (CD27+) B lymphocytes.
Defects of the “intrinsic” (mitochondrial) pathway of apoptosis leading to ALPS include gain-of-function mutations of NRAS and somatic mutations of KRAS , but these variants do not result in an increased number of DNTs.
Management of ALPS includes use of immunosuppressive drugs (steroids, mycophenolate mofetil, sirolimus) to control autoimmune manifestations and regular monitoring of lymphoproliferation by computed tomography scanning or positron emission tomography for surveillance of lymphoma. Splenectomy may improve the cytopenias but is associated with a marked increase of sepsis. Chronic cytopenias tend to improve over time; HSCT should be reserved for very severe clinical presentations with autoimmune disease refractory to treatment.
ITCH Deficiency
Mutations of the E3 ubiquitin ligase ITCH have been shown to cause multisystem autoimmune disease in affected individuals from a large Amish kindred. Characteristic clinical features include failure to thrive, hepatosplenomegaly, multiorgan autoimmune manifestations (hypothyroidism, hepatitis, diabetes, enteropathy), chronic lung disease, developmental delay, dysmorphic features, and hypotonia. Dysfunction of E3 ubiquitin ligases have been shown to lead to indiscriminate T cell activation, loss of tolerance to self-antigens and accumulation of autoreactive B lymphocytes.
Immunodeficiencies with Impaired Cell-Mediated Cytotoxicity
CD8+ cytotoxic T lymphocytes (CTLs) and NK cells play a critical role in antiviral immune defense through two mechanisms: Fas-mediated apoptosis of virus-infected cells and release of cytolytic granules. In resting CTLs and NK lymphocytes, cytotoxic proteins are contained in endosomal secretory granules. On activation of CTLs and NK lymphocytes, lytic granules polarize toward the immune synapse, dock, and fuse with the cell membrane of the effector cell. Multimers of perforin form pores through which CTLs and NK cells release cytotoxic proteins (granzyme B, granulolysin) into virus-infected target cells, causing activation of caspases and apoptosis of target cells.
Defects in this process are responsible for various forms of familial hemophagocytic lymphohistiocytosis. Defective cell-mediated cytotoxicity is also observed in several forms of pigmentary dilution disorders (discussed previously). Finally, selective susceptibility to EBV infection is at the basis of various forms of lymphoproliferative syndrome. These diseases and their associated genes are listed in Table 24-6 .
| Syndrome | Gene |
|---|---|
| Familial hemophagocytic lymphohistiocytosis | PRF1, STX11, STXBP2, UNC13D |
| X-linked lymphoproliferative syndromes | BIRC4, SH2D1A |
| Other | CD16, CD27, ITK |
Familial Hemophagocytic Lymphohistiocytosis
Familial hemophagocytic lymphohistiocytosis (FHL) includes a heterogeneous group of monogenic disorders characterized by impaired cell-mediated cytotoxicity. In patients with FHL, the inability to clear viral infections causes persistent activation of CD8+ and NK lymphocytes, with production of high amounts of IFN-α and -β and of proinflammatory cytokines (tumor necrosis factor-alpha [TNF-α], IL-6). Clinical features of FHL include recurrent episodes of high fever, pancytopenia, liver and spleen enlargement, and signs of neurologic involvement. The diagnostic criteria were revised in 2007. Elevated serum levels of triglycerides, ferritin, and soluble CD25 and decreased levels of fibrinogen and poor NK function are typical laboratory findings. If left untreated, FHL is rapidly fatal. Severe bacterial and fungal infections and multiple organ failure are the main causes of death. Treatment should be aimed at eradicating the underlying infection while suppressing immune activation at the same time. Corticosteroids, cyclosporine A, etoposide, and antithymocyte globulin are often effective in tempering immune activation. However, relapses are common, and the only curative treatment is represented by HSCT. In particular, reduced intensity conditioning has shown excellent efficacy and limited toxicity.
The genetic defect has been identified for four forms of FHL so far, and all of them are inherited as autosomal recessive traits. Perforin deficiency accounts for approximately 30% of all cases of FHL. It is caused by mutations of the PRF1 gene that prevent expression or correct assembly of perforin multimers that form pores through which cytolytic enzymes are released into target cells. Mutations of the UNC13D gene, encoding for Munc13-4, impair priming of the cytolytic granules, which is necessary for their fusion with the cell membrane. UNC13D mutations represent the most common form of FHL (30% to 40% of all cases). Defects of the Syntaxin 11 (STX11) gene affect granule exocytosis. Finally, FHL may also be caused by defects of Munc18-2, a protein partner of STX11 encoded by the Syntaxin binding protein 2 (STXBP2) gene.
X-Linked Lymphoproliferative Disease
X-linked lymphoproliferative (XLP) disease is characterized by unique susceptibility to severe complications after EBV infection in male subjects. Two genetically distinct forms are known. In XLP1 the disease is caused by mutations of the SH2 domain containing protein 1 A (SH2D1A) gene, encoding for a small adapter protein (SLAM-associated protein, or SAP) involved in intracellular signaling in T and NK lymphocytes. In particular, SAP binds to CD244 (a SLAM family member) expressed by NK lymphocytes. EBV-infected B cells express high levels of CD48, the CD244 ligand. On CD244/CD48 binding, SAP participates at intracellular signaling and promotes NK cytolytic activity against EBV-infected target cells. In the absence of SAP, NK cytolytic activity is impaired. SAP is also important for the function of follicular helper T cells that promote maturation of antibody responses. XLP2 is caused by mutations of the BIRC4 gene, which encodes for the X-linked inhibitor of apoptosis (XIAP) protein.
XLP1 and XLP2 have partially overlapping clinical features. Hemophagocytic lymphohistiocytosis (HLH) subsequent to primary EBV infection is a prominent sign at clinical presentation in both diseases. XLP1 may manifest with fatal infectious mononucleosis, aplastic anemia, pulmonary lymphoid granulomatosis with vasculitis, and lymphoma. A proportion of XLP1 patients who survive primary EBV infection will develop hypogammaglobulinemia, which in some cases is associated with elevated levels of IgM or IgA. Some patients may have a clinical and laboratory phenotype consistent with common variable immunodeficiency (CVID, discussed later). Functional activity of NK and CTLs is impaired. Finally, patients with XLP1 lack NKT lymphocytes. Without HSCT, 62.5% of the patients survive (and most of them require Ig replacement); however, poor survival rates (less than 20%) have been observed in patients without transplants who have features of HLH. HSCT may cure approximately 80% of XLP1 patients, but poorer outcome (50% survival) has been observed if HLH is present.
In contrast, XLP2 causes recurrent episodes of HLH, with or without EBV infection. Patients with XLP2 are not at increased risk of lymphoma; on the other hand, inflammatory bowel disease is frequently observed. Definitive treatment of XLP2 is also based on HSCT; however, use of a myeloablative conditioning regimen is associated with high mortality.
IL-2 Inducible Tyrosine Kinase Deficiency
Interleukin-2 inducible tyrosine kinase (ITK) is a nonreceptor tyrosine kinase that modulates the strength of intracellular signaling in T lymphocytes on TCR-induced activation. Patients with ITK deficiency are susceptible to EBV-driven lymphoproliferative disease (often involving the lungs as well), Hodgkin lymphoma, hypogammaglobulinemia, autoimmunity, and HLH. The disease is inherited as an autosomal recessive trait. There is a reduced number of naïve CD4+ lymphocytes and of NKT cells. Allogeneic HSCT may cure the disease.
CD27 Deficiency
CD27 is a member of the TNF receptor family and is broadly expressed on the surface of various lymphocyte subsets. By interacting with CD70, it promotes generation and maintenance of T-cell memory. It is also commonly used as a marker of memory B lymphocytes. CD27 deficiency has been identified in some patients presenting with severe EBV infection. Lymphoma and secondary hypogammaglobulinemia have been reported in some of these patients. HSCT represents a curative form of treatment.
CD16 Deficiency
One of the mechanisms by which NK lymphocytes mediate killing of target cells is antibody-dependent cellular cytotoxicity (ADCC), whereby antibody-bound target cells come in contact with NK lymphocytes through binding of the Fc region of IgG molecules to a specific receptor (FcγRIIIa, also known as CD16) expressed on the surface of NK lymphocytes. Homozygosity for the Leu66His amino acid substitution in CD16 causes deficient spontaneous NK cell-mediated cytotoxicity but surprisingly leaves ADCC intact. Expression of CD2, a co-activating NK cell receptor, is decreased, affecting intracellular signaling in NK lymphocytes. Recurrent and severe herpesvirus infections are the clinical hallmark of this condition.
Immunodeficiencies with Selective Predisposition to Pathogens
Most forms of primary immunodeficiencies are characterized by susceptibility to a broad range of pathogens, whose nature (bacteria, viruses, fungi, protozoa) reflects the underlying immune defect. However, in the last two decades it has become clear that certain gene defects that affect the immune system may lead to selective predisposition to a narrow group of pathogens. These findings have supported the genetic theory of infectious diseases, whereby life-threatening infections early in life may in fact be caused by inborn errors of immunity. These disorders and their associated genes are listed in Table 24-7 .
| Syndrome | Gene |
|---|---|
| Mendelian susceptibility to mycobacterial disease | IFNGR1, IFNGR2, IL12B, IL12RB1, STAT1 |
| Epidermodysplasia verruciformis | TMC6, TMC8 |
| Trypanosomiasis | APOL1 |
Mendelian Susceptibility to Mycobacterial Diseases
The designation Mendelian susceptibility to mycobacterial diseases (MSMD) refers to a group of PIDDs characterized mainly by selective susceptibility to infection with mycobacteria including both Mycobacterium tuberculosis and many atypical species. Patients are also susceptible to disseminated infections with Salmonella species. There are no other consistent clinical manifestations, and all routine screening tests of humoral and cellular immunity are normal. The vast majority of these patients harbor mutations in one of five genes that encode elements in a signaling cascade involving IFN-γ and IL-12. IL-12 is produced by mononuclear cells in response to stimulation by way of TLRs and other pattern recognition receptors. IL-12 stimulates T h 1 differentiation, and mature T h 1 cells and NK cells produce IFN-γ. IFN-γ then acts on mononuclear cells to promote intracellular killing of phagocytosed organisms. In the absence of the normal operation of this sequence, macrophages are not able to kill ingested mycobacteria or Salmonella organisms, and severe infection results. Thus far, mutations have been found in the genes encoding the IL-12 p40 subunit (IL12B), the β1 chain of the IL-12 receptor (IL12RB1), both chains of the IFN-γ receptor ( IFNGR1, IFNGR2), and the STAT1 signal transducing molecule which mediates signaling of the IFN-γ receptor. Most cases show autosomal recessive inheritance, but a few specific mutations of IFNGR1 may exhibit autosomal dominant inheritance. Acquired forms of susceptibility to mycobacterial infection have been ascribed to autoantibodies to IFN-γ.
Therapy for MSMD is mainly through aggressive treatment of infections when they occur and prophylaxis for selected individuals who may relapse frequently. For patients with hypomorphic (partly functional) mutations of IFNGR1, IFN-γ supplementation may be beneficial.
Epidermodysplasia Verruciformis
Mutations in the genes TMC6 (transmembrane channel-like 6) and TMC8 give rise to epidermodysplasia verruciformis (EV). These genes were previously called EVER1 and EVER2 , respectively. EV exhibits autosomal recessive inheritance, and affected individuals are susceptible to persistent skin infection only by HPV. There is a high rate of malignant transformation in the skin, but there are no other infectious susceptibilities or clinical manifestations of the disease. Acquired EV has been associated with human immunodeficiency virus (HIV) infection.
Herpes Simplex Encephalitis
HSVs are prevalent in the general population and are capable of causing disseminated severe and even fatal infections in patients with profound T-cell defects (discussed earlier). However, in rare cases individuals are susceptible specifically to encephalitis caused by HSV, in the absence of any other organ or system involvement. To date, mutations in five distinct genes have been associated with this clinical entity: TLR3 (TLR3), UNC93B1, TRAF3, TICAM1 (previously known as TRIF ) , and TBK1 . All of the proteins encoded by these pathways interact in transducing responses by way of TLR3 (and in some cases also TLR4) with the common result of a failure to produce sufficient amounts of type 1 interferons in response to HSV infection in the brain. Inheritance varies according to the genes and the specific mutations involved. Thus far, mutations in UNC93B1 have shown exclusively autosomal recessive inheritance, whereas mutations of TRAF3 and TBK1 have exhibited exclusively autosomal dominant inheritance. Mutations of TLR3 and TICAM1 have shown either dominant or recessive inheritance depending on the specific lesion.
Susceptibility to Trypanosomiasis
Trypanosomes are single-cell (protozoan) organisms causing endemic infections in many parts of the world. Trypanosoma cruzi is the causative agent of Chagas disease, and it is estimated that approximately 10 million people in South America harbor this parasite, most being asymptomatic. Subspecies of Trypanosoma brucei are responsible for sleeping sickness, and it is estimated that more than 60 million individuals in Africa harbor this organism. Most individuals are protected from progression of disease by apolipoprotein L1 (APOL1), which can lyse the T. brucei plasma membrane and kill the organism. Some strains of trypanosomes have become more pathogenic by virtue of their resistance to APOL1 lysis. Conversely, individuals who have mutations in their APOL1 genes are more susceptible to trypanosome infection and severe disease. There are no known clinical consequences of deleterious APOL1 mutations beyond susceptibility to T. brucei. However, isoforms of APOL1 selected through evolution for higher trypanosome lytic activity are risk factors for nondiabetic kidney disease in African-Americans.
Humoral Immunodeficiency
The humoral immunodeficiencies and their associated gene defects are listed in Table 24-8 . These disorders are characterized by hypogammaglobulinemia or dysgammaglobulinemia, often together with relative deficiency of antibody responses to various forms of antigenic challenge. Cellular immunity is generally intact. Humoral immunodeficiency is most commonly manifested by recurrent bacterial and viral infections of the upper and lower respiratory tract. Pyogenic skin infections, as well as meningitis, osteomyelitis, and other foci of infection, are also seen frequently. These infections are generally caused by the same organisms that are virulent in immunocompetent hosts, predominantly encapsulated bacteria such as Streptococcus pneumoniae , Hemophilus influenzae, S. aureus , and Neisseria meningitidis . Viral infections usually resolve normally in these patients, although a higher rate of recurrence with the same agent may be seen because there is impaired production of neutralizing antibodies or B-cell memory. The results of physical examination are not specific and show only the presence or sequelae of microbial infections. A relative paucity of peripheral lymphoid tissue may be noted in some patients, especially areas rich in B cells (e.g., tonsils). Studies of peripheral blood lymphocyte subsets are often normal; the number of B cells may be reduced in certain syndromes.
| Syndrome | Gene |
|---|---|
| X-linked (Bruton) agammaglobulinemia | BTK |
| Autosomal recessive agammaglobulinemias | CD79A, CD79B, BLNK, IGHM, IGLL1, LRRC8, PIK3R2 |
| Monogenic common variable immunodeficiency | BAFFR, CD19, CD21, CD81, ICOS |
| Common variable immunodeficiency | Undefined |
| Good syndrome | Undefined |
| Selective IgA deficiency | Undefined |
| IgG subclass deficiency | Undefined |
| Specific antibody deficiency with normal immunoglobulins | Undefined |
| Transient hypogammaglobulinemia of infancy | Undefined |
| Class-switch defects: | |
| CD40 ligand deficiency (X-linked HIM) | TNFSF5 |
| CD40 deficiency | TNFRSF5 |
| B-cell–specific defects | AICDA, UNG |
The Agammaglobulinemias
X-Linked Agammaglobulinemia
X-Linked agammaglobulinemia (XLA) is also known as Bruton agammaglobulinemia because Bruton provided the classic description of the disease in 1952. Affected male subjects are often asymptomatic during the first months of life while they are protected by transplacentally acquired maternal antibodies. These antibodies fall to low levels by 6 to 9 months of age, and patients begin to experience recurrent bacterial infections. Physical examination at any age often reveals a striking absence of tonsils and palpable lymphoid tissue. Serum Ig is absent or the level is extremely low; in addition, no B cells are found or their numbers are extremely low. Cellular immunity is completely intact. However, these patients are prone to the development of chronic enteroviral meningoencephalitis and vaccine-associated paralytic poliomyelitis, which suggests an important role for humoral immunity in the control of these infections. Moreover, patients may have arthropathy resembling rheumatic disease, which may be due to infection with Mycoplasma or Ureaplasma organisms. Other opportunistic infections, such as P. jiroveci pneumonia, are seen only rarely.
XLA is another example of an immunodeficiency caused by a defect in a signal transduction molecule. In fact, this protein tyrosine kinase is now known as Bruton tyrosine kinase ([Btk], Fig. 24-7 ). It is expressed in B cells at all stages of development, as well as in cell lines derived from monocytes, macrophages, mast cells, and erythroid cells. A variety of BTK gene defects such as point mutations, deletions, and insertions have been described in patients with XLA. In most patients B-cell development is impeded at an early stage (the pro–B-cell to pre–B-cell transition).