PRIMARY HYPOGONADISM
Part of “CHAPTER 115 – MALE HYPOGONADISM“
Including infertile men, primary testicular failure affects 5% to 10% of the male population.4 Although male infertility is commonly considered a problem that involves exclusively seminiferous tubular function and spermatogenesis, evidence from men with varicoceles has demonstrated the presence of an exaggerated response of both serum LH and FSH to LHRH.6 Although total serum testosterone levels are normal in these patients, the augmented gonadotropin response to LHRH indicates a failure in both Leydig cell and seminiferous tubular function (Fig. 115-1). Some compensation for the impaired Leydig cells must occur that is sufficient to return testosterone levels to normal under the influence of increased LH stimulation. These findings confirm that infertile men often possess primary gonadal failure of a subtle nature. Because the incidence of male infertility is fairly high, primary testicular dysfunction in the male population is an important issue.
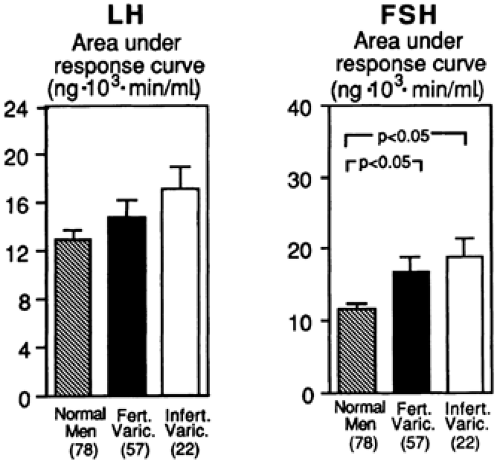 FIGURE 115-1. Exaggerated luteinizing hormone (LH) and follicle-stimulating hormone (FSH) response to gonadotropin-releasing hormone in fertile and infertile men with a varicocele is compared with that of normal men. Results suggest impairment of both seminiferous tubule and Leydig cell function. Bars indicate standard error of the mean. (Fert. Varic., fertile men with varicocele; Infert. Varic., infertile men with varicocele.) (From Nagao RR, Plymate SR, Berger RE, et al. Comparison of gonadal function between fertile and infertile men with varicoceles. Fertil Steril 1986; 46:930.) |
KLINEFELTER SYNDROME
The chromosomal constitution of 47,XXY epitomizes the classic form of male primary testicular failure. This abnormality is present in ˜1 in 400 men. Klinefelter syndrome was first described in 1942 in nine male patients who, at puberty, experienced the onset of bilateral gynecomastia, small testes with Leydig cell dysfunction and azoospermia, and increased urinary gonadotropin excretion.7 Later, Leydig cell failure was shown to be variable in its magnitude. In 1956, the X chromatin body (Barr body) was found in these individuals, and in 1959, the XXY chromosome constitution was first described, demonstrating that the disease was the result of an extra X chromosome (see Chap. 90).8,9
PHENOTYPIC MANIFESTATIONS
The phenotypic manifestations of Klinefelter syndrome are characteristic for the classic form of the disease in which all cells carry the XXY karyotype. Many men with Klinefelter syndrome have a mosaic form, in which some cell lines are XXY and others are XY. In these mosaic individuals, all cell lines are from a single zygote, and the XXY cell lines arise from mitotic nondisjunction after fertilization. Manifestation of the disease may not be typical or consistent. Before puberty, the only physical findings are the small testes; in the classic form of Klinefelter syndrome, a gonadal volume of <1.5 mL after the age of 6 years is usual.10,11 Regardless of the patient’s age, a decreased testicular size is the most diagnostic clinical feature of classic Klinefelter syndrome. A suggested cause for the small prepubertal testicular size is a loss of germ cells before puberty; thus, the prepubertal testis is small. In secondary forms of hypogonadism, the number of germ cells is normal, and the prepubertal testicular size is indistinguishable from that of normal boys.
Microcephaly also has been described in some cases of Klinefelter syndrome. After puberty, the more characteristic features of the syndrome appear (Fig. 115-2 and Fig. 115-3). These include varying degrees of gynecomastia. The gynecomastia is of interest because it is due primarily to an increase in periductal tissue rather than ductal tissue; increased ductal tissue often is found in gynecomastia of acute onset, whatever the cause. If the gynecomastia were due only to the increased estrogen production in Klinefelter syndrome, an increase in ductal tissue might be expected. Nonetheless, the histopathologic appearance of the breast tissue is not consistently separable from that of other patients with long-standing gynecomastia.
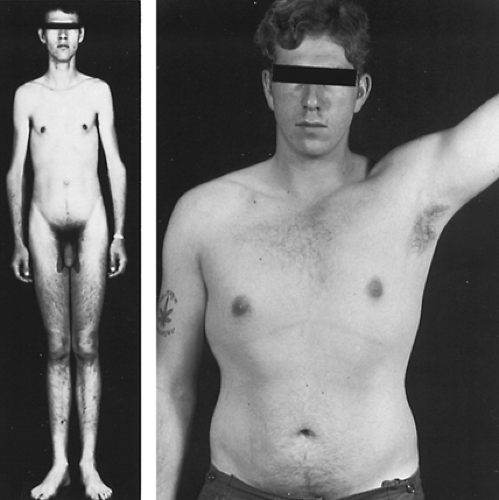 FIGURE 115-2. Two patients with Klinefelter syndrome. The man on the left has the phenotypic features of a markedly increased lower body segment. He has XXY/XY mosaicism in all tissues, including the testes. On semen analysis, his sperm count was 300,000/mL. The patient on the right has moderate gynecomastia, but a relatively normal masculine appearance. He has an XXY karyotype. |
 FIGURE 115-3. Patient with Klinefelter syndrome before and after treatment with testosterone. He had a sparsity of body hair and marked gynecomastia. Note the diminution of subcutaneous fat, the increased body hair, and the increased muscle mass after therapy. In addition, the patient had breast tissue removed. (From Becker KL. Clinical and therapeutic experiences with Klinefelter syndrome. Fertil Steril 1972; 23:568.) |
After puberty, an abnormality in skeletal proportions becomes manifest. The lower extremities show exaggerated growth, resulting in a decreased crown-to-pubis/pubis-to-floor ratio, such as that seen in eunuchoid men (see Chap. 18). However, unlike in the true eunuchoid state, in which the arm span often is at least 6 cm more than the height, in Klinefelter syndrome, the arm span/height ratio usually is not abnormal. The reason for the abnormal growth in the lower extremities is unknown. The phenotypic manifestations, such as the gynecomastia, pattern of hair distribution, muscle mass, and subcutaneous fat distribution, vary even among patients with classic Klinefelter syndrome.
LABORATORY FINDINGS
In patients with Klinefelter syndrome, serum testosterone levels range from low to well into the normal range12,13 (Fig. 115-4). The
relatively decreased serum testosterone is the result of deficient testosterone production (3.27 ± 1.35 mg per 24 hours vs. 7.04 ± 2.47 mg per 24 hours in normal persons).14 An increase in SHBG also occurs in Klinefelter syndrome; thus, the bioavailable, or free, testosterone is further suppressed.12,15 As men with Klinefelter syndrome age, a further decline in testicular function often occurs.16
relatively decreased serum testosterone is the result of deficient testosterone production (3.27 ± 1.35 mg per 24 hours vs. 7.04 ± 2.47 mg per 24 hours in normal persons).14 An increase in SHBG also occurs in Klinefelter syndrome; thus, the bioavailable, or free, testosterone is further suppressed.12,15 As men with Klinefelter syndrome age, a further decline in testicular function often occurs.16
 FIGURE 115-4. Changes in serum gonadotropin and testosterone levels in boys with Klinefelter syndrome as they are followed up through puberty. Note the lack of differentiation from normal values until after puberty has supervened. (LH, luteinizing hormone; FSH, follicle-stimulating hormone.) The numbers represent actual values for three patients whose values (77, 48, and 68) exceeded the scale of the graph. (From Ratcliffe SG. Klinefelter’s syndrome in children: a longitudinal study of 47 XXY boys identified by population screening in Klinefelter’s syndrome. In: Bandmann HJ, Breit R, Perwein E, eds. Klinefelter’s syndrome. New York: Springer-Verlag, 1984:38.) |
Patients with Klinefelter syndrome may have serum estradiol levels higher than those of normal men. The increase in estradiol has been shown to come from an augmented peripheral conversion of testosterone to estradiol.12,14 After puberty, serum gonadotropin levels are elevated, even when serum testosterone levels are normal.13 Before puberty, serum gonadotropin levels are
normal17 (see Fig. 115-4). Interestingly, in spite of the relatively normal masculine appearance of some young men with Klinefelter syndrome, their physical strength often is significantly less than that of their peers because of the deficient androgen action on muscle. Men with Klinefelter syndrome usually do not have the ability to grow a full beard or a mustache; however, like muscle strength, androgen-dependent hair growth is variable.
normal17 (see Fig. 115-4). Interestingly, in spite of the relatively normal masculine appearance of some young men with Klinefelter syndrome, their physical strength often is significantly less than that of their peers because of the deficient androgen action on muscle. Men with Klinefelter syndrome usually do not have the ability to grow a full beard or a mustache; however, like muscle strength, androgen-dependent hair growth is variable.
The abnormal serum testosterone levels and the diminished response of serum testosterone to human chorionic gonadotropin (hCG) is a reflection of the disturbance of Leydig and Sertoli cell function. Of the two compartments in the testes, the seminiferous tubules are the most affected by the presence of the extra X chromosome.18,19 and 20 Clinically, this is manifested by azoospermia, which occurs in virtually all patients with classic Klinefelter syndrome. Testicular biopsy specimens consistently demonstrate seminiferous tubule hyalinization and fibrosis (Fig. 115-5). Because the seminiferous tubule basement membrane is so crucial to spermatogenesis, the defect in basement membrane formation may explain why the initial testicular damage is to the seminiferous epithelium.
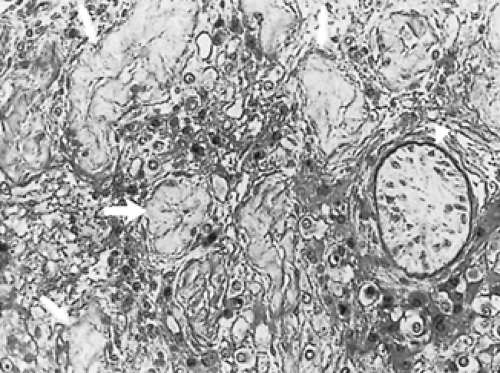 FIGURE 115-5. Testis biopsy specimen from a patient with XXY Klinefelter syndrome demonstrating hyalinized tubules (arrows) with focal areas of immature seminiferous tubules (arrowhead). |
In some patients with Klinefelter syndrome, usually the mosaic forms, testicular biopsy specimens have demonstrated focal areas of spermatogenesis. On occasion, sperm have been noted in the ejaculate, and these patients may be fertile. However, most of the reported impregnations have been single events occurring early in the individual’s reproductive life, with azoospermia developing in later years.21 Is it the lack of normal androgen environment in the testes or the chromosomal abnormality that impairs spermato-genesis? In part, the answer to this question has come from studies of XXY and XX sex-reversed mice in which sperm can develop but are always diploid, with an XX or XY content.22 In these animals, during spermatogonial mitosis, the XY and XX daughter cells are not viable, but an occasional nondisjunctional event takes place and the extra chromosome is lost. Thus, in subsections of the germinal epithelium, a normal haploid germ cell develops and full spermatogenesis may take place. The testicular biopsy specimens of these animals, as well as of patients with Klinefelter syndrome, show focal areas of spermatogenesis. Interestingly, the XXY fetal testis may be unaffected.23 Meiotic segregation of sperm nuclei has been evaluated in sperm obtained from a patient with Klinefelter syndrome; by fluorescence in situ hybridization analysis, 958 cells contained the X chromosome and 1077 cells contained the Y chromosome. This ratio is significantly different from the 1:1 ratio that was expected (chi2 test, p < .01). The frequency of 24,XX, 24,XY, and diploid cells was significantly increased. These results suggest that the 47,XXY cells are able to complete meiosis and produce mature sperm nuclei.24
PSYCHOPATHOLOGY
Decreased intellectual development and antisocial behavior occur with a high frequency in Klinefelter syndrome.25,26 and 27 In this regard, the incidence of Klinefelter syndrome in subsets of the population with mental retardation ranges from ˜0.5% to 2.5%. Studies from Denmark, in which the records of all men taller than 184 cm were surveyed, show that the incidence of criminal offense was 9.3% in men with an XY chromosome pattern, 18.8% in men with an XXY chromosome pattern, and 41.7% in men with an XYY chromosome pattern.28 Furthermore, the incidence of criminal behavior was inversely related to the subjects’ full intelligence quotient (IQ). Studies of boys with Klinefelter syndrome have found that a significant reduction in verbal IQ is present at the age of 7 years.29 Interestingly, as the number of X chromosomes increases in the poly-X disorders, the degree of mental retardation also increases. The mechanism by which the extra X chromosome impairs central nervous system (CNS) function has not been delineated, although an inverse relationship exists between the ability to cope and antisocial behavior. Furthermore, although androgen production by the Leydig cells is decreased, low androgen production does not appear to be responsible for all the characteristic clinical manifestations of the syndrome (other states of androgen deficiency are not necessarily associated with increased criminal behavior and mental
retardation). The extra X chromosome may affect neuronal function directly, which then leads to behavioral abnormalities related to decreased IQ and other, nondefined factors.
retardation). The extra X chromosome may affect neuronal function directly, which then leads to behavioral abnormalities related to decreased IQ and other, nondefined factors.
Other areas of psychiatric dysfunction, such as personality traits of timidity, introspective behavior, and social drive, are more clearly related to the androgen deficiency and often improve after androgen replacement.30 The lack of self-restraint and aggressiveness shown by some subjects appears to be related to the effects of the extra X chromosome on CNS function.
ETIOLOGY
The development of the XXY chromosome constitution may occur by several mechanisms. The most common of these is nondisjunction during the first and second meiotic division in parental oogenesis or spermatogenesis (see Chap. 90). A second possibility for the development of the classic syndrome is nondisjunction during the first postzygotic mitosis. Nondisjunction in mitosis after this time results in mosaic forms of Klinefelter syndrome. A third, and rare, mechanism is anaphase lag during mitosis or meiosis, with the lagging chromatid included in the daughter nucleus. Reports have been published of six men with an XXY chromosome pattern (including one set of brothers) whose mothers had XX/XXX chromosomes.9 These family data suggest a nondisjunctional event during maternal zygotic development, and this may have been present in a grandparent. Because Klinefelter syndrome usually is not recognized until after puberty, most mothers of affected men have not had chromosome analyses performed.
The reason for the nondisjunction is not known, although maternal age has been shown to be a factor. The incidence of Klinefelter syndrome increases from 0.6% when the maternal age is 35 years or less to 5.4% when the maternal age is >45 years.31,32 Sixty-five percent of patients with Klinefelter syndrome have a maternal origin for their extra X chromosomes. Maternal age is thought to be a factor because of the longer diplotene stage of the ova in older women (see Chap. 94). The existence of mothers with an XX/XXX chromosome pattern (see earlier) and an increased incidence of twins with Klinefelter syndrome, as well as the finding of an increased frequency of Klinefelter syndrome in patients with Down syndrome, suggest that some families may have a factor predisposing to chromosomal nondisjunction.
DISEASE ASSOCIATIONS AND MEDICAL COMPLICATIONS
Numerous disease associations have been made with Kline-felter syndrome; especially prominent are malignancies and autoimmune diseases. Malignancies include breast carcinoma (with an incidence 20 times greater than that of men with an XY chromosome pattern and 20% that of women), nonlymphocytic leukemia, lymphomas, marrow dysplastic syndromes, and extragonadal germ cell neoplasms.33,34,35,36 and 37 Autoimmune diseases, especially chronic lymphocytic thyroiditis and rare syndromes such as Takayasu arteritis, have been reported in Klinefelter syndrome.38,39,40,41 and 42 Whether this results from an effect of decreased androgens on the OKT4/OKT8 lymphocyte ratios or from the combined effect of decreased immune surveillance and increased X chromosome material is unknown.43,44
Taurodontism (enlargement of the molar teeth by an extension of the pulp) is present in 40% of men with Klinefelter syndrome, compared with ˜1% of men with an XY chromosome pattern.45 Varicose veins, with or without hypostatic ulceration, are seen in 40%; this, along with the 55% incidence of mitral valve prolapse, suggests a connective tissue defect.46,47 Abnormal glucose tolerance due to a postreceptor defect in insulin action is seen in 10% of patients with Klinefelter syndrome.48 The incidence of asthma and chronic bronchitis is also increased. Although osteoporosis is increased, the frequency is no greater than in other male hypogonadal states.49,50,51 and 52
As with most hypogonadal syndromes, sexual activity or sexual orientation is of concern to these patients and their family members or spouses. The patients with marked hypogonadism tend to associate less in adolescence with male peer groups because of their physical inability to compete. However, patients with Klinefelter syndrome do not have any greater homosexual tendencies than do men with an XY chromosome pattern. Usually, heterosexual activity is less frequent among patients with Klinefelter syndrome; many do not attain orgasm during sexual activity.53
VARIANT FORMS
In certain patients, the sex chromosome configuration may show two or more stem cell lines (mosaicism). For example, in a testicular biopsy prepared for chromosome analysis, the predominant cell line may be XXY but another line of normal XY cells also may be present. Thus, on metaphase analysis, an XXY/XY cell pattern is found. This lack of a pure XXY cell line may result in a modification of the phenotypic characteristics of the patient such that the only clinical manifestation of classic Klinefelter syndrome is infertility (Table 115-3). On the other hand, if most cells are XXY, the patient may appear to have the classic Klinefelter syndrome, yet because of a normal XY cell line in some testicular germ cells, spermatogenesis and fertility may occur54 (see Fig. 115-5). Approximately 10% of patients with Klinefelter syndrome have a mosaic chromosomal constitution.
 |
The existence of mosaic forms may be suspected in men who are infertile with small testes or who have primary hypogonadism and normal buccal smears. In these patients, if more than one tissue specimen is examined for the presence of an XXY chromosome constitution (e.g., leukocytes and testicular tissues), and an extra X chromosome is found in one cell line but not the other, evidence for mosaicism exists.
Importantly, the examination for chromatin in tissues often is poorly performed and may lead to erroneous conclusions. For example, many laboratories allow “normal” men to have an occasional X-chromatin body in a buccal smear. These are not true X-chromatin bodies but folds in cells (or bacteria). Therefore, when Klinefelter syndrome, especially the mosaic form, is suspected, confirmation should be made by a formal karyotyping (Fig. 115-6).
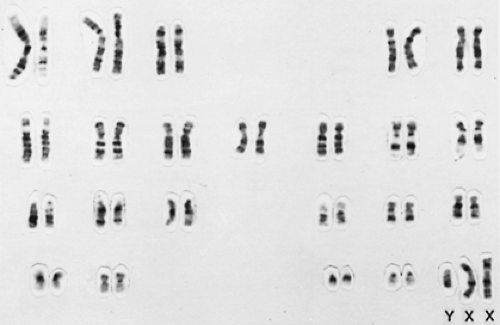 FIGURE 115-6. Karyotype with Giemsa banding from a peripheral lymphocyte of a patient with Klinefelter syndrome. Note the similar banding patterns of the duplicated X chromosome. (Courtesy of C. M. Disteche, Department of Pathology, University of Washington, Seattle, WA.) |
Several additional variant forms of Klinefelter syndrome have been reported (see Table 115-3). These usually arise from consecutive nondisjunctional events in oogenesis or spermato-genesis. Individuals with the poly-X chromosomal syndromes (e.g., XXXY, XXXXY) display more severe abnormalities than do those with the classic Klinefelter syndrome55 (Fig. 115-7). A clue to these disorders is the presence of more than one chromatin body on buccal smear preparations. Often, marked mental retardation is present. The seminiferous tubules may have undergone hyalinization before puberty, and the testes commonly are undescended. Skeletal abnormalities, including proximal radioulnar synostosis and overgrowth of the radioulnar head, are characteristic of these disorders. As evidence of more severe androgen deficiency, the development of the genitalia may be retarded and a bifid scrotum or hypoplastic penis may be present.
 FIGURE 115-7. Youth with the XXXXY syndrome. Epicanthal folds, a hypoplastic midface, and prognathism are seen. The neck is short, the penis is small, and the scrotum is hypoplastic. (From Goodman RM, Gorlin RJ. The face in genetic disorders. St. Louis: CV Mosby, 1977:722.)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|