Chapter Outline
PLATELET MORPHOLOGY AND SUBCELLULAR ORGANIZATION
PLATELET GENOMICS AND PROTEOMICS
ANTITHROMBOTIC COMPONENTS OF THE VESSEL WALL
PLATELET GRANULES AND THEIR ROLE IN HEMOSTASIS
FEED-FORWARD AMPLIFICATION PATHWAYS INVOLVED IN PLATELET RECRUITMENT AND THROMBUS STABILITY
CELL SURFACE AND CYTOSOLIC PROTEINS THAT LIMIT PLATELET RESPONSIVENESS
In an 1881 communication to the Turin Royal Academy of Medicine, the Italian physician Giulio Bizzozero disclosed the presence in circulating human blood of discrete elements that he termed “ piastrine ” (“ Blutplättchen ” in a 1882 publication in a German journal and “ petites plaques ” in a communication in French). Previously speculated to be merely nonphysiologic “granular aggregates,” blood platelets have since become central to the understanding of thrombosis and hemostasis, and detailed understanding of their participation in cardiovascular disease, stroke, and even cancer has led to remarkable progress in the rational treatment of these disorders.
Although platelets are most often studied in the context of their ability to form a hemostatically effective plug, it is now widely recognized that their influence extends far beyond this process to all aspects of hemostasis, as well as to wound healing and vascular remodeling. For example, platelets generate or secrete biologically active mediators such as thromboxane A 2 (TXA 2 ) and serotonin, which not only amplify platelet activation responses but also modulate vascular tone. In addition, platelets secrete a broad array of granule constituents that stimulate vessel repair, induce megakaryocytopoiesis, promote coagulation, and limit fibrinolysis.
The same pathways that lead to platelet plug formation can also produce pathologic thrombosis, a process that has been described as hemostasis occurring at the wrong time or in the wrong place. Platelets are particularly important for hemostasis on the arterial side of the circulation, where blood flows under higher pressure and experiences greater shear force. As a result, platelet function is generally considered to be critical to the pathogenesis of arterial thrombosis and less so for venous thrombosis, and antiplatelet drugs are most widely used in the former setting. However, this distinction between the mechanisms underlying arterial and venous thrombosis is not absolute, and the spectrum of thrombotic disorders should be considered a continuum.
Arterial thrombosis is a particularly common problem in middle-aged and older adults and is a major cause of morbidity and mortality in developed countries. The thrombi that arise in atherosclerotic vessels are predominantly platelet in origin and are the proximate cause of myocardial infarction and most cerebrovascular accidents. Although arterial thrombosis is considerably less common in children than adults, it may contribute to major morbidity in patients with sickle cell disease, as well as complications of some childhood infections, Kawasaki syndrome, and various forms of arteritis, autoimmune disorders, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura (see Chapter 34 ).
In this chapter platelet structure and function are reviewed, and special emphasis is given to the cell surface glycoproteins (GPs) that function as sentries for areas of vascular damage and the signal transduction events that both amplify and limit platelet responsiveness. The information provided should be helpful in understanding subsequent chapters that describe inherited and acquired platelet disorders (see Chapters 30 and 34 ) and the role of the adhesive protein von Willebrand factor (VWF) in hemostasis (see Chapter 31 ). Finally, there is growing appreciation of the role that platelets play in inflammation and the pathogenesis of atherothrombosis, which is briefly discussed at the end of the chapter.
Platelet Morphology and Subcellular Organization
Platelets are adhesion and signaling machines that circulate as small, disc-shaped cellular fragments in the whole blood of healthy individuals at a concentration of approximately 150,000 to 300,000 per µL. Early studies suggested that platelets might be produced via cytoplasmic fragmentation along a network of internal demarcation membranes that were observed in large, polyploid megakaryocytes. More recent studies, however, support the notion that proplatelets are assembled and packaged with their various constituents at the ends of long cytoplasmic extensions of differentiated megakaryocytes that have migrated from the proliferative osteoblastic niche to the capillary-rich vascular niche of the bone marrow microenvironment, with the invaginated demarcation membrane system serving simply as a reservoir of internal membrane used for proplatelet extension. Once adjacent to the adluminal face of the endothelium, proplatelets are released into the blood stream, where they circulate as mature platelets for approximately 7 to 10 days before being cleared by the liver and spleen, their life span being controlled at least in part by an antagonistic balance between the apoptotic proteins Bcl-xL and Bak.
The size of resting platelets is somewhat variable, averaging approximately 1.5 µm in diameter and 0.5 to 1 µm in thickness. Platelet size is undoubtedly regulated by numerous factors during their biogenesis, but both the 224-kDa nonmuscle myosin heavy chain IIA (MYHIIA) and the cell-surface glycoprotein Ib complex (GPIb) appear to play critical roles. Thus mutations in the MYH9 gene act dominantly to interfere with contractile events important for platelet formation, whereas failure to express GPIb results in Bernard-Soulier syndrome by disruption of critical associations with the cytoskeletal protein filamin, which play an essential role both in platelet formation and in platelet compliance. In both of these inherited platelet disorders, platelets are as large as lymphocytes (see Chapter 30 ). Correction of GPIb expression in GP Ib-deficient (Bernard-Soulier syndrome) mice has been shown to restore platelets to their normal size.
The volume of platelets (mean platelet volume) normally ranges from 6 to 10 femtoliters. Platelet density is also variable, and the issue of whether young platelets are more or less dense as they gain versus lose content during their circulating lifetime has never been satisfactorily resolved. Because platelets retain most species of messenger ribonucleic acid (mRNA) for a short period after their release from bone marrow megakaryocytes, young platelets can be distinguished from older ones based on their RNA content.
As shown in Figure 26-1, A and C , resting platelets are discoid in shape, largely because of the presence of a circumferential coil of microtubules, and are packed with numerous electron-opaque α granules, a few dense (δ) granules, several mitochondria, and lysosomes. Platelets also retain a few Golgi bodies, as well as occasional vestiges of rough endoplasmic reticulum—the exception being platelets from patients with rapid platelet turnover, where very young platelets containing more abundant protein synthesis machinery are readily observed in the circulation. Platelets also contain two highly specialized membrane systems not found in other cells of the body: the surface-connected open canalicular system (OCS; see Fig. 26-1, B and C ), and the dense tubular system (DTS). The OCS is a series of tortuous invaginations of the plasma membrane that appear to tunnel throughout the cytoplasm of the cell that serves as an internal reservoir of plasma membrane that is called upon when platelets round up, extend lamellipods and filopods (see Fig. 26-1, B and D ), and spread during platelet activation—a process that can increase by more than 400% the surface area of exposed plasma membrane. Because OCS channels are proximal to internal granules, they also likely function as a conduit for the rapid expulsion of α- and δ-granule contents during platelet activation. The DTS, on the other hand, is a remnant of the smooth endoplasmic reticulum and is found randomly dispersed throughout the cytoplasm. The DTS appears to be one of several organelles within the platelet known to harbor high concentrations of calcium and is thought to contain a 100-kDa calcium adenosinetriphosphatase (ATPase) known as sarcoplasmic/endoplasmic reticulum Ca 2+ -ATPase 2b ( SERCA2-b) that functions to sequester and store cytosolic calcium in resting cells. Recent evidence suggests that adenosine diphosphate (ADP) is able to induce selective release of calcium from the DTS, whereas activation of the GPIb/V/IX receptor for VWF releases calcium primarily from a poorly defined acidic compartment within the cell. Thrombin, a strong platelet agonist, appears to elicit calcium release from both stores upon binding to the platelet thrombin receptor PAR-1.
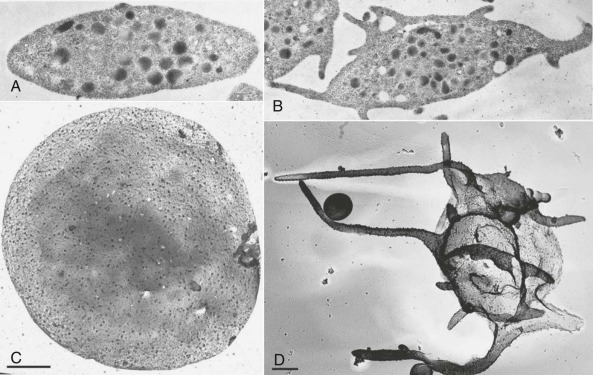
The platelet cytoskeleton is comprised of a single, rigid but dynamic microtubule of approximately 100 µm in length coiled 8 to 12 times around the equatorial plane of the cell. This marginal band of microtubules is largely responsible for maintaining the discoid shape of the resting cell, as illustrated by the observations that 1) incubation of platelets with colchicine, an agent that dissolves microtubules, results in their rounding ; 2) platelets from mice lacking β 1 tubulin remain largely spherical ; and 3) “platelet spherocytosis” in humans results when tubulin fails to polymerize normally into microtubules. Directly underneath the plasma membrane lies an intricate, two-dimensional, tightly-woven membrane skeleton comprised of nonerythroid spectrin, a network of actin filaments, vinculin, and the actin-binding protein filamin, which itself is tethered to the inner face of the plasma membrane via linkages with the cytoplasmic domain of GPIb. The membrane skeleton, because of its location, serves as a scaffold, linking elements of the plasma membrane with contractile elements of the cytoskeleton and cytosolic signaling proteins, and thereby regulates such diverse functions as receptor mobility, receptor clustering, and signal transduction. Finally, the platelet is filled with an extensive cytoplasmic network of actin filaments organized by the actin-binding proteins filamin and α-actinin that constitute its cytoskeleton.
When platelets become exposed to components of the extracellular matrix or to soluble agonists like ADP or thrombin, they undergo dramatic changes in their morphology. The marginal band of microtubules disappears, allowing the platelet to transform from a disc to an irregular sphere. At nearly the same time, the actin filament capping protein, α-adducin, becomes phosphorylated and dissociates from existing F-actin filaments, exposing the barbed end of the filament to cytosolic actin monomers and driving rapid polymerization of actin into microfilaments. This has the dual effect of driving the extension of lamellipodia and filopodia, and of forcing granules towards the center of the platelet where they can fuse with membranes of the OCS and release their contents to the exterior of the cell. Phosphorylation of myosin additionally induces contractile events that facilitate centralization of the granules.
Platelet Genomics and Proteomics
Though anucleate, platelets contain measurable and manipulable levels of megakaryocyte-derived mRNA, at least some of which is capable of being synthesized into small but detectable amounts of protein. Both serial analysis of gene expression (SAGE) and gene microarray analyses were employed early on to estimate the size and composition of the platelet transcriptome. Of the 20,563 genes present in the human genome, 9538 distinct transcripts (6493 in the mouse) were identified in unstimulated platelets using RNA sequencing (RNA-SEQ). That is considerably fewer than normally found in a nucleated cell, but perhaps more than might have been expected from an anucleate circulating cellular fragment. One of the more surprising findings in recent years has been the identification of heavy (sometimes called heterogeneous ) nuclear RNA (hnRNA) in the platelet cytosol, as well as all of the spliceosome components necessary to splice the hnRNA into mature message that can be thereafter translated into protein. Enlisted during the activation process, signal-dependent protein translation has thus far been demonstrated for mRNAs encoding interleukin (IL) 1β, tissue factor, and Bcl-3, the protein products of which have the potential to influence inflammation, thrombosis, and wound repair. Platelets also contain an abundance of microRNAs (miRs) that have the potential, especially in precursor megakaryocytes, to regulate transcription and translation.
The platelet proteome appears to be equally complex and diverse, and unlike the transcriptome, reports both the breadth as well as the relative amounts of protein products actually present in the cell. Obtained by refined two-dimensional gel electrophoretic techniques that were originally developed in the 1970s or by liquid chromatographic separation, proteins are fragmented and separated using a combination of proteolytic and ionization techniques and then analyzed by mass spectrometry, allowing for the identification of thousands of proteins present in complex cellular lysates or subcellular fractions. In by far the most complete and quantitative profiling analyses to date, Burkhart and colleagues have identified a core platelet proteome comprised of approximately 5000 distinct platelet proteins, including an abundance of molecules involved in adhesion, signaling, cytoskeletal change, and metabolism. Others have combined prefractionation methods with suitable separation techniques to describe the inventory of platelet proteins that become phosphorylated during platelet activation, can be palmitoylated, or are enriched within various subcellular compartments such as the platelet cytoskeleton, platelet α-granules, the membrane fraction, membrane rafts, and microparticles.
Antithrombotic Components of the Vessel Wall
Though hundreds of thousands of platelets per microliter circulate in blood, very few, if any, interact with the intact vessel wall, because the endothelial lining of the blood vessel presents an excellent nonthrombogenic surface. In fact, this property of the vessel wall has not yet been duplicated in any prosthetic or extracorporeal device. The healthy endothelium not only provides an effective barrier between blood and the highly thrombogenic components of the subendothelium (see below), but also actively produces both membrane-bound and secretory products that limit fibrin generation and promote clot dissolution. For example, heparinlike glycosaminoglycans present on the luminal side of the endothelial cell surface recruit plasma antithrombin, effecting a conformational change that promotes binding and neutralization of thrombin and other serine proteases. Thrombin, when bound to the endothelial cell surface receptor, thrombomodulin, takes on anticoagulant properties via its cleavage and activation of protein C, which in turn cleaves coagulation factors V and VIII, suppressing further thrombin generation. Endothelial cells also express a specific receptor for activated protein C, which serves to concentrate the protein on the endothelial surface. Finally, endothelial cells synthesize, secrete and rebind tissue plasminogen activator, which activates plasminogen to facilitate fibrin dissolution ( Fig. 26-2 ).
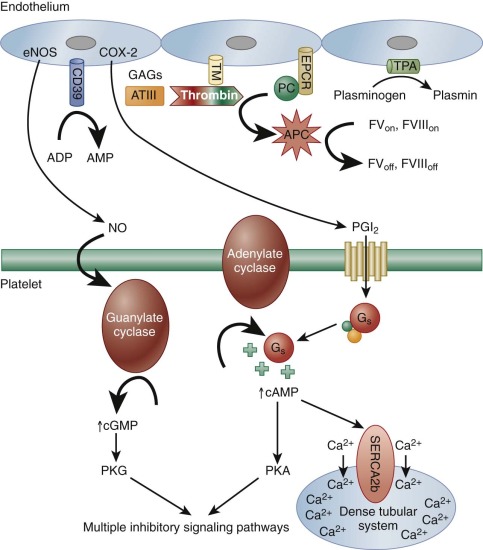
The endothelial cell also produces two important inhibitors of platelet activation: prostacyclin (PGI 2 ) and nitric oxide (NO). The latter was first recognized for its role as an endothelium-derived relaxation factor. A labile oxygenated metabolite of arachidonic acid generated by endothelial cell cyclooxygenase-2 (COX-2), PGI 2 diffuses out of the cell and binds to a platelet G s -protein coupled receptor (GPCR) known as the isoprostenoid (IP) receptor. Its release stimulates adenylate cyclase to increase cytosolic cyclic adenosine monophosphate (cAMP) levels, which 1) activates a pump in the DTS that decreases cytosolic calcium ions (Ca 2+ ), thus helping keep platelets quiescent, and 2) activates protein kinase A (PKA). PGI 2 also has potent vasodilatory effects, binding to IP on arterial smooth muscles cells to effect vessel relaxation. PGI 2 produced by the vascular endothelium thus serves to counterbalance the proaggregatory and vasoconstrictor activities of the platelet-derived prostanoid, TXA 2 , the biology of which is discussed below. In fact, upsetting the delicate balance between COX-1–derived TXA 2 and COX-2–derived PGI 2 has been shown to increase the risk of adverse cardiovascular events.
Whereas PGI 2 stimulates adenylate cyclase to produce cAMP, NO, a product of L-arginine generated by endothelial nitric oxide synthase (eNOS), directly activates platelet guanylate cyclase, resulting in increased levels of cytosolic cyclic guanosine monophosphate (cGMP). Although platelet responses to low levels of this cyclic nucleotide can at first be mildly stimulatory, cGMP, largely via its activation of protein kinase G (PKG), has the overall effect of dampening platelet responses and inhibiting platelet adhesion, platelet aggregation, and platelet-mediated recruitment of leukocytes during the inflammatory response (see Fig. 26-2 ). In addition to these two soluble metabolites, endothelial cells also express on their surface a potent ADPase known at CD39, which scavenges plasma ADP to prevent platelet aggregation. Finally, it is important to note that inflammatory cytokines, oxidized lipids, and immune complexes can, under pathologic conditions, inhibit these protective biochemical pathways and impair the antithrombotic state of the endothelial cell. These changes permit untoward formation of platelet- and fibrin-containing thrombi, as well as thrombus formation beyond sites of vascular injury, and can thus contribute to atherothrombosis.
Reacting to the Breach: Cell Surface Receptors That Mediate Tethering and Adhesion and Transmit Early Activation Signals
As antithrombotic as is the endothelial lining, the underlying extracellular matrix consists of a rich mixture of glycosaminoglycans into which are embedded an abundance of highly-concentrated prothrombotic proteins, including structural components like collagen and elastin (which comprise ≈30% of body weight) and adhesive proteins such as laminin, fibronectin, and VWF. Not surprisingly, platelets have evolved receptors for most of these proteins, and a series of rapid biochemical events is initiated both on the surface and inside the cell when exposed to them. Adhesion is an activating event.
The large number of circulating red blood cells serves to marginate platelets, and when the vessel wall is breached, either by mechanical injury or after atherosclerotic plaque rupture, the first layer of platelets to encounter exposed matrix undergoes a series of sequential events similar to that which leukocytes experience during the inflammatory response—namely tethering, initial signaling to the cell interior, integrin-mediated adhesion, and cytoskeletally-directed cell spreading. Whereas leukocyte tethering is mediated by members of the selectin family, the first layer of platelets become tethered on VWF, which is sprinkled on exposed collagen fibers via interactions with a high-affinity, platelet-specific, multi-subunit receptor known as the GPIb/V/IX complex. This complex, which is expressed at approximately 25,000 copies/platelet, binds to the A1 domain of VWF with sufficiently high affinity to tether platelets even under conditions of arterial shear. Loss of this receptor in both humans and mice results in a clinical condition known as Bernard-Soulier syndrome, which is characterized not only by an increase in platelet size but also by prolonged bleeding caused, in large part, by the inability of platelets to adhere to the vessel wall. After engagement with its ligand, GPIb acts through membrane-proximal Src family kinases, adapter molecules, and to a lesser extent via its association with immunoreceptor tyrosine-based activation motif (ITAM)–bearing subunits to transmit early activation signals. Together the result is the recruitment and activation via tyrosine phosphorylation of phospholipase Cγ2 (PLCγ2), a key enzymatic component of platelet amplification that is required to achieve thrombus growth and stability ( Figure 26-3 ).
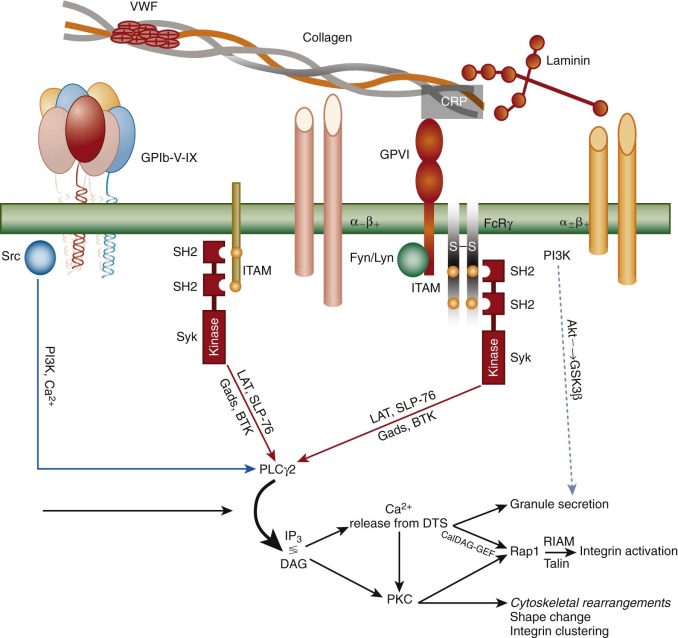
Once tethered, two different platelet integrins—each of which exists in a low-affinity state on the platelet surface—begin to engage specific extracellular matrix components, and together with the small calcium transients and kinase-generated signals emanating from the GPIb complex, and from mechanical shear forces generated by the flowing blood, initiate the reciprocal processes of platelet adhesion and activation. Thus the integrin α 2 β 1 binds to exposed collagen fibrils, and the integrin receptor α 6 β 1 engages laminin. Both of these integrins “hand off” to a member of the immunoglobulin superfamily, glycoprotein VI (GPVI), which via its non–covalent association in the plane of the plasma membrane with the ITAM-bearing Fc-receptor γ-chain homodimer, elicits strong PLCγ2-dependent events that 1) begin the process of cytoskeletally-directed shape change and cell spreading; 2) initiate signal transduction pathways that cause dramatic structural changes in platelet integrins (see Fig. 26-3 ), resulting in their adopting a high-affinity, ligand-binding–competent conformation (a process known as “inside-out” signal transduction); and 3) facilitate fusion of α- and δ-granules with the OCS and underlying plasma membrane. Other cell-surface receptors that participate in adhesion-initiated activation events include PEAR1, FcγRIIa, and the C-type lectin receptor, CLEC-2.
Platelet Granules and Their Role in Hemostasis
Platelet-specific granules are synthesized, assembled, and packaged during megakaryocyte biogenesis, and at later stages of maturation they appear to come into contact with microtubules. The microtubules then transport them, via the microtubule motor protein kinesin, along the shafts of proplatelets until they reach the proplatelet tips. Once inside a mature platelet, platelet granules remain relatively evenly dispersed throughout the cytoplasm, their contents awaiting threshold signals for cellular activation, at which time their membranes fuse with those of the plasma membrane, or more likely, the invaginated subdomains of the OCS. The regulated secretion of granule contents ensures that hemostasis remains highly localized, an event that has been exploited to deliver non–platelet-derived procoagulant proteins like Factor VIII to sites of vascular injury.
Platelets harbor three distinct types of granules ( Box 26-1 ). Two, α-granules and dense (δ) granules, are found only in platelets, whereas lysosomes are present in nearly all cell types. α-Granules are by far the most numerous, with as many as 40 to 80 per cell, and they contain a wide array of proteins and bioactive peptides. Box 26-1 classifies α-granule proteins as those that reside within the α-granule membrane (P-selectin being the most diagnostic), those pinocytosed from plasma and packaged (IgG, fibrinogen, albumin), and those synthesized by megakaryocytes and stored (VWF, platelet factor 4, thrombospondin). mRNAs encoding the latter group have all been identified in the platelet cytoplasm. Upon platelet activation, granules redistribute toward the center of the cell, at which time soluble N -ethylmaleimide–sensitive attachment protein receptor (SNARE) proteins within the α-granule membrane facilitate fusion with members of the Rab family of low–molecular-weight GTPases playing a prominent role in vesicle docking and exocytosis. After membrane fusion, P-selectin and α-granule membrane-specific proteins like TLT-1 become expressed on the platelet surface, and the contents of the granule are released into the plasma milieu. Exposed P-selectin, diagnostic of an activated platelet, serves to recruit leukocytes to the site of injury, constituting one of a number of important links between thrombosis and inflammation. Proteins secreted from the platelet include the adhesive ligands VWF and fibrinogen, which serve to support platelet–platelet interactions, growth factors and cytokines that promote cell migration, wound healing, maintenance of vascular integrity, and autocrine factors like growth-arrest–specific gene 6 (Gas6) and CD40L that are released and rebind platelet receptors to help amplify platelet responsiveness. Finally, α-granules and their contents are a source of procoagulant proteins, releasing Factor V and exposing tissue factor to promote localized fibrin deposition at sites of vascular injury. Platelet α-granules, or at least their contents, are severely reduced in an inherited bleeding disorder known as the gray platelet syndrome (GPS) (see Chapter 30 ).
- A.
α-Granules
- 1.
Membrane proteins enriched in the granule membrane: P-selectin, TLT-1, CD40 ligand (which is cleaved after exposure on the platelet surface to release soluble CD40L), and tissue factor.
- 2.
Membrane proteins present at similar concentrations as they are in the plasma membrane: GPIIb-IIIa, GPIb, PECAM-1, and perhaps many others
- 3.
Granule contents:
- a.
Synthesized by megakaryocytes: thrombospondin, VWF, PF4, β-thromboglobulin, PDGF
- b.
Endocytosed from plasma or origin not determined: albumin, fibrinogen, fibronectin, IgG, Gas6, coagulation factor V, and many chemokines and growth factors, including RANTES, bFGF, EGF, TGFβ, and VEGF
- a.
- 1.
- B.
Dense (δ) granules: ADP, ATP, 5′-HT, Ca ++ , polyphosphate
- C.
Lysosomes: acid hydrolases, elastase, cathepsins, and other degradative enzymes
5′-HT, 5′-hydroxytryptamine; ADP, adenosine diphosphate; ATP, adenosine triphosphate; Ca 2+ , calcium ion; EGF, epidermal growth factor; FGF, fibroblast growth factor; IgG, immunoglobulin G; PDGF, platelet-derived growth factor; PF4, platelet factor 4; RANTES, regulated on activation, normal T-cell expressed and secreted; TGF, transforming growth factor; VFW, von Willebrand factor.
Dense granules (four to eight per platelet) are morphologically distinct, electron-opaque storage organelles that contain the vasoconstrictive substance serotonin, adenine and guanine nucleotides like ADP and ATP, inorganic pyrophosphates and polyphosphates, and Ca 2+ . Complexes of the latter two are likely responsible for the dark appearance of these bodies in thin section electron microscopy. Dense granule membranes contain a few components in common with lysosomal membranes, like granulophysin (CD63, lysosomal associated membrane protein [LAMP]-3) and LAMP2, as well as membrane proteins also present in α-granule membranes, like P-selectin, suggesting a common origin during biogenesis. Like α-granule membrane proteins, these become expressed on the platelet-cell surface after granule fusion and secretion and can be used as platelet activation markers. Curiously, a number of plasma membrane GPs, including GPIb and GPIIb-IIIa, have also been reported in dense granule membrane. Dense granule contents, especially ADP, play a physiologically important role in hemostasis, as evidenced by characteristic platelet function defects in patients whose platelets lack dense granules or their contents, collectively known as storage pool disorders. Chédiak-Higashi and Hermansky-Pudlak syndromes are two such examples of autosomal recessive dense granule defects that lead to platelet dysfunction and bleeding, the former being associated with immunodeficiency and the latter with albinism (see Chapter 30 ).
Primary lysosomes are the third organelle whose contents are secreted upon platelet activation, though only three or fewer per cell are normally identifiable. Though a clear role for lysosomes in platelet function has not been identified, they do contain more than a dozen different acid hydrolases, cathepsins D and E, and other degradative enzymes that can be secreted if platelets are subjected to strong agonist stimulation. Their contents have been shown to be mildly reduced in the platelets of individuals with GPS, in keeping with the notion that GPS is caused by a defect in packaging. Membrane proteins on platelet lysosomes are typical of lysosomes in other cells, and include LAMP-1, LAMP-2, and LAMP-3.
Feed-Forward Amplification Pathways Involved in Platelet Recruitment and Thrombus Stability
Although platelet adhesion, early activation signals, and granule release are necessary prerequisites for thrombus formation, efficient recruitment of additional platelets to the site of the vascular lesion to yield a stable platelet plug requires a host of additional receptor/ligand interactions, each of which results in signal transmission and therefore biochemical and cell biological changes that help sustain platelet activation. Among the most important of these is the binding of released ADP to one of its two platelet G-protein–coupled receptors, P2Y 12 . Like the α 2 receptor for epinephrine, P2Y 12 is coupled to an inhibitory G protein that slows down the activity of adenylate cyclase, thus lowering cytosolic levels of cAMP ( Fig. 26-4 ). This greatly potentiates platelet responses to other agonists, because it allows calcium ions (released from the DTS as a result of the action of phospholipase-generated 1,4,5-inositol trisphosphate [IP 3 ]) to remain cytosolic. “Bioavailable” calcium ions in turn support a host of additional cellular events, including more robust granule secretion, activation of metal ion-dependent proteases, and activation of cell surface integrins. The importance of ADP in amplifying platelet responses is illustrated by the clinical effectiveness of ticlopidine and clopidogrel—widely-used pharmacological agents that antagonize the activity of P2Y 12 —in pacifying platelet reactivity and inhibiting platelet aggregation.
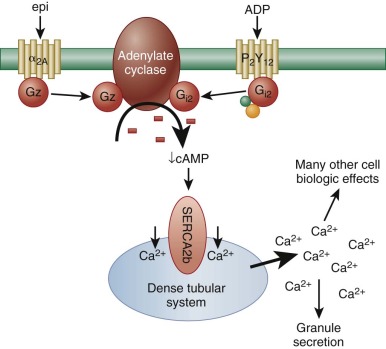
In addition to ADP-induced, P2Y 12 -mediated signaling, there are nearly a dozen other soluble ligands that are either generated or released at sites of vascular injury and that function in signal amplification and platelet activation. These can be broken into three classes according to the type of platelet receptor to which they bind ( Fig. 26-5 ). The first class of ligands is comprised of ADP, thrombin, TXA 2 , and serotonin (5′-hydroxytryptamine), each of which bind to a specific GPCR that is coupled to the heterotrimeric a subunit, Gq. Thus ADP binds to P2Y 1, thrombin to the protease activated receptors PAR1 and PAR4, TXA 2 to the thromboxane receptor, and serotonin to 5HT2A. When released as a consequence of ligand binding to any of these GPCRs, the Gq subunit binds to the β isoform of phospholipase C (PLC). PLCs are lipid hydrolases that act on membrane-associated phosphatidylinositol (PI)–4,5-P 2 to produce the second messengers IP 3 and diacylglycerol (DAG). IP 3 binds and opens calcium channels, and DAG activates the most abundant forms of protein kinase C (PKC), initiating additional signaling cascades downstream of this serine/threonine kinase. As shown in Figure 26-5 , it is the sum of these products—generated by the γ2 isoform of PLC in response to adhesion and by the β isoform of PLC in response to ligand-GPCR interactions—that the platelet integrates when deciding whether to become fully activated. This concept is important in the context of designing pharmacologic strategies to inhibit platelet function, because blocking adhesion and its consequent activation of PLCγ2 leaves PLC β-mediated platelet activation largely intact and vice versa.
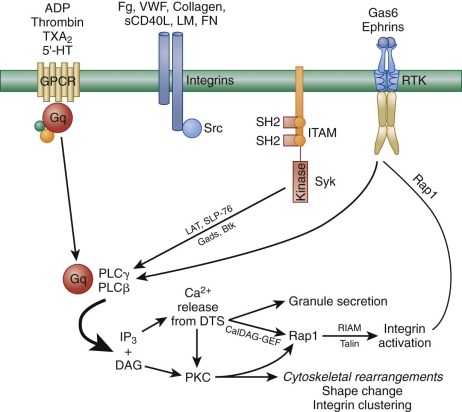
The second class of signal amplifiers is the cell surface integrins themselves. As illustrated in the middle section of Figure 26-5 , when ligands bind to integrins, Src-family kinases (SFKs) associated with integrin cytoplasmic tails trigger a series of incompletely understood SFK-mediated amplification events that have been collectively termed outside-in signaling. This has recently been demonstrated after interaction of the major platelet integrin αIbβ 3 with its ligand fibrinogen; however, signals also emanate from α 2 β 1 and α 6 β 1 when platelets encounter collagen and laminin at sites of vascular injury. Activation signals from the latter two may be relatively weak by comparison, owing to the fact that only a few thousand of each are expressed on each platelet, compared with 50 to 80 thousand αIbβ 3 receptors per cell. Protein kinases Syk and FAK have been shown to become activated downstream of αIIbβ 3 engagement, as has PLCγ 2. However, the details of these events remain to be worked out. A less well-defined autocrine loop that utilizes integrin-mediated outside-in signal amplification is the cleavage and rebinding of soluble CD40L after α-granule fusion and secretion.
The third class of feed-forward amplification reactions is mediated by ligand-activated plasma membrane receptor tyrosine kinases (RTKs). The first of these to be described were receptors for Gas6, a vitamin K-dependent protein related to the anticoagulant protein S. Gas6 is thought to reside in platelet α-granules, and like other α-granule proteins, becomes secreted upon platelet activation. Interestingly, platelets have three different receptors for Gas6: Axl, Sky, and Mer, all of which have active cytoplasmic tyrosine kinase activity (see Fig. 26-5 ). Upon engagement, Gas6 receptors trigger tyrosine phosphorylation of the β 3 integrin cytoplasmic domain and thereby support outside-in integrin signaling, as well as activate PI3-kinase to further sustain granule secretion. Platelets also express two members of the Eph receptor tyrosine kinase family, EphA4 and EphB1, which when in contact with their membrane-bound counter-receptor, Ephrin B1, stimulate tyrosine phosphorylation of the integrin β 3 tail and activate the integrin activator, Rap1b. Like Gas6 signaling, genetic loss or pharmacologic blockade of Ephrin/Eph kinase interactions results in decreased ability to form a stable thrombus or retract a fibrin clot.
Activation of the Major Platelet Integrin, αIIbβ 3 (GPIIb-IIIa Complex): the Final Common Endpoint of Platelet Activation.
Human platelets express at least five different members of the 24-member integrin family, including three β1 integrins (α 2 β 1 , α 5 β 1 , and α 6 β 1 —specific for collagen, fibronectin, and laminin, respectively) and two β 3 integrins—avβ 3 and its close relative, αIIbβ 3 (also known as the GPIIb-IIIa complex). αIIbβ 3 is by far the most abundant and well-studied. This section will focus on our current understanding of how αIIbβ 3 becomes transformed from a resting to an active, ligand binding-competent conformation, with the understanding that the biochemical and cell biological principles described for this integrin may well apply to the others.
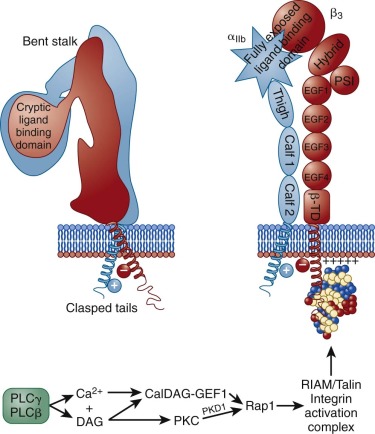
As shown in schematic form on the left side of Figure 26-6 , αIIbβ 3 exists on the platelet surface in a bent-over conformation that is unable to associate effectively with its major soluble ligands, fibrinogen, VWF, and fibronectin. Though relatively short, the cytoplasmic domains of αIIb and β 3 are thought to play a key role in maintaining the “off” state of this integrin complex, owing to weak charge interactions between them that allow the hydrophobic transmembrane domain helices of each subunit to interact and maintain the integrin in a low-affinity state. When platelets become activated—either by adhesion– or soluble-agonist–mediated events—calcium and DAG, generated as a result of the actions of PLCγ2 and PLCβ (see Figs. 26-3 and 26-5 ), bind to and activate PKC and the guanine exchange factor, CalDAG-GEF1. As illustrated in Figure 26-6 , each of these can independently activate Rap1, a low molecular weight GTPase that has been implicated in integrin activation. Rap1 activates integrins via an effector molecule known as the Rap1 GTP -interacting adaptor molecule (RIAM), which recruits the highly abundant cytosolic protein, talin to the inner face of the plasma membrane, forming an integrin activation complex. Binding of RIAM-associated talin to the β 3 integrin subunit represents the final common step in integrin activation, because it disrupts the weak ionic clasp between the αIIbβ 3 tails, allowing for tail separation and a dramatic, rapid unfolding of the extracellular domain. Simultaneous conformational changes in the integrin head result in formation of an integrin receptor having high affinity for its soluble adhesive ligands. In a still-to-be-defined mechanism, the cytoskeletal protein kindlin 3 then contributes to activating and/or maintaining the activation state of αIIbβ 3 . As integrin-rich adhesion complexes mature, the cytoskeletal protein vinculin takes over for the nascent Rap1/RIAM complex and stabilizes adhesions. Finally, integrins cluster, ensuring that bound ligands effectively broker with high avidity the platelet–platelet interactions that permit thrombus growth and stabilization.
Cell Surface and Cytosolic Proteins That Limit Platelet Responsiveness
As anyone who has witnessed a myocardial infarction or thrombotic stroke will know, unrestrained thrombus growth at inappropriate sites can be as harmful as excessive bleeding, because it can result in vessel occlusion, ischemia, and tissue damage. Numerous active processes are in place to limit platelet responsiveness in healthy vessels, to keep thrombus growth localized to specific sites of vascular injury, and to facilitate dissolution of a platelet plug during recovery.
Exogenous inhibitors of platelet function produced by a healthy endothelial lining contribute to platelet passivation via continuous generation of PGI 2 and NO, which act on platelets by activating adenylate and guanylate cyclases to increase intracellular levels of cAMP and cGMP, subsequently activating PKA and PKG (see Fig. 26-2 ). PKG controls the threshold for platelet activation primarily by phosphorylating the IP 3 receptor-associated cGMP (IRAG) kinase substrate, a protein that associates with PKG and IP 3 receptor type I to inhibit IP 3 -induced calcium release from intracellular stores. Both PKA and PKG interfere with platelet activation by phosphorylating and inactivating vasodilator-stimulated phosphoprotein (VASP), a molecule with anticapping activity that is important for the processes of actin polymerization and filopod formation. PKCδ also binds VASP and interferes with its ability to promote filopodia formation, although this pathway is unique to collagen-stimulated platelets and does not involve regulation of PKA- or PKG-mediated VASP phosphorylation.
In addition to exogenous inhibitors, platelets themselves contain a plethora of inhibitory proteins designed to keep platelet activation in check, either by setting a threshold for platelet activation or by turning off activation signals once sufficient platelet responses have occurred. Regulator of G-protein signaling (RGS) proteins modulate GPCR-mediated platelet accumulation and thrombus formation at sites of vascular injury, as do members of the β-arrestin family. Platelets also express three members of the immunoglobulin gene–immunoreceptor tyrosine-based inhibitory motif (Ig-ITIM) family of inhibitory receptors, PECAM-1, CEACAM-1, and G6b-B, each of which contains in its cytoplasmic domain tyrosine residues that, when phosphorylated form docking sites for phosphatases that function to shut down tyrosine kinase–dependent signaling reactions. Interestingly, one of the five tyrosine kinases present in platelets, Lyn, actually limits platelet aggregation both in response to GPVI-specific stimuli and after platelet spreading on immobilized fibrinogen, perhaps via its ability to phosphorylate the ITIM tyrosines of PECAM-1.
Several inhibitory pathways have been identified in platelets that either regulate, or are regulated by PI3 kinase, a lipid kinase that phosphorylates the 3′ position of PI 4,5-diphosphate (PIP 2 ) to generate PI 3,4,5-triphoshate (PIP 3 ), thereby creating docking sites on the inner face of the plasma membrane for pleckstrin homology (PH) domain–containing molecules. The actions of PI3-kinase are opposed by the lipid phosphatase, SH2 domain-containing inositol 5′-phosphatase 1 (SHIP1), which hydrolyzes the 5′-phosphate of PIP 3 . In platelets, SHIP1 has been shown to downregulate PIP 3 generation after αIIbβ 3 -mediated outside-in signaling, and thus it may interfere with feed-forward amplification pathways that increase the efficiency with which platelets are recruited to growing thrombi. Interestingly, the 5′-inositol phosphatase activity of SHIP1 appears to be enhanced at least in part by the actions of the Src-family tyrosine kinase,
Akt (also known as protein kinase B ) is a PH domain–containing serine/threonine kinase that is a well-characterized effector of PI3 kinase. Akt contributes positively to platelet activation in multiple ways, one of which appears to be by inactivating the serine/threonine kinase, glycogen synthase kinase 3 (GSK3). The GSK3 family is comprised of three isoforms (α, β, β 2 ) that are constitutively active in resting cells but which become inactivated in activated cells by Akt-mediated phosphorylation. Platelets express two isoforms of GSK3 (α and β), both of which become phosphorylated and inactivated after exposure of the platelet to multiple agonists that activate PI3 kinase and Akt. Whereas initial studies reported that specific inhibitors of GSK3 activity block rather than enhance platelet responses to agonist stimulation, more recent data implicate the β isoform of GSK3 as a negative regulator of platelet function both in vitro and in vivo. Finally, the serine/threonine kinase PKCδ has also been shown to negatively regulate platelet activation after platelet exposure to collagen.
Nonhemostatic Roles for Platelets: Blood Vessel Maintenance and Repair, Inflammation and Infection, and Atherothrombosis
In addition to being essential for primary hemostasis, activated platelets and their secreted products have the ability to influence a broad array of pathophysiologic processes. These include leukocyte trafficking and inflammation, tissue regeneration and angiogenesis, and both the beginning and end stages of atherosclerosis.
Activated platelets that become spread on components of the extracellular matrix or on each other display an altered surface phenotype, the most prominent of which is exposure of several thousand copies of the α-granule-derived membrane protein, P-selectin. P-selectin is also expressed on cytokine-activated endothelial cells. Thus after either a thrombotic or inflammatory event, P-selectin appears on the luminal face of the vessel wall, where it serves to recruit monocytes and neutrophils into the underlying tissue by binding PSGL-1, a constitutively expressed counter-receptor for P-selectin that is present on most leukocytes. In vivo, mice lacking P-selectin exhibit greatly diminished leukocyte rolling, delayed recruitment into sites of inflammation, and increased susceptibility to infection. Whereas endothelial P-selectin no doubt has a major role in leukocyte capture, platelet P-selectin likely plays a prominent role in “secondary capture.” As in platelets, tethering also initiates activation of leukocyte integrins, which are then able to mediate cell spreading and transendothelial migration. P-selectin–PSGL-1 interactions therefore constitute an important link between thrombosis and inflammation. Other platelet/leukocyte receptor–counter-receptor pairs have also been shown to facilitate the inflammatory response, including binding of platelet-associated fibrinogen binding to the leukocyte integrin, MAC-1 and platelet JAM-3 binding to MAC-1 on monocytes and dendritic cells.
In addition to forming a platform for leukocyte recruitment during acute inflammation, platelets also deliver to the vessel wall proinflammatory chemokines that are thought to play roles in the development of atherosclerosis by promoting further chemoattraction of leukocytes and by stimulating proliferation of vessel wall smooth muscle cells and fibroblasts. Such secreted factors include the C-X-C chemokine platelet factor 4, macrophage inflammatory protein (MIP) 1a, the C-C chemokine regulated on activation, normal T-cell expressed and secreted (RANTES), CD40 ligand, platelet-derived growth factor (PDGF), and TGFβ. Activated platelets also synthesize de novo IL 1β, a potent stimulator of endothelial cells and monocytes that upregulates adhesion molecule expression. Thus platelets appear to contribute in a number of ways to the development and progression of atherosclerotic lesions.
So as not to leave the reader with the impression that platelets only exacerbate chronic human disease, it should be noted that as early as 1969 platelets and their secreted products were shown to be able to “nurture” the vascular endothelium. They are increasingly used as a source of biological response modifiers in a plethora of conditions, including organ preservation, gum restoration after dental procedures, and tissue repair after surgery. The ability of platelets to adhere to sites of vascular injury and secrete both degradative enzymes and at the same time growth-promoting factors like vascular endothelial growth factor (VEGF), PDGF, fibroblast growth factor (FGF), epidermal growth factor (EGF) and angiopoietin 1 allows them to play a uniquely supportive role in endothelial cell migration and survival during the process of wound healing and angiogenesis. Platelet-rich plasma to treat bone and cartilage repair after sports injuries, though largely unsubstantiated, is also finding increasing use. Platelet-secreted factors have also been shown to have a critical role during the time when embryonic blood vessels and lymphatic vessels separate during development in a mechanism that involves binding of the ITAM-bearing platelet receptor CLEC-2 to the lymphatic endothelial marker podoplanin. Blood flow also appears to be a critical regulator of this developmental event. Platelet secreted products also play a vital role in another developmental event, namely closing of the ductus arteriosis. Finally, platelets bind via their cell-surface CD36 receptor to malarial-infected red blood cells, an event that induces secretion of platelet factor 4 (PF4) from platelet α granules. PF4 in turn binds to the Duffy antigen on the red blood cells, causing lyses of the parasite digestive vacuole, ultimately killing the parasites within.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


