34 Pituitary Tumors
Pituitary adenomas account for 25% of all intracranial tumors and are the most common lesion arising in the sellar region.1 They are present in approximately 16.9% of the general population.2 Although men and women are equally affected, certain tumor subtypes do show a sex preference. Pituitary tumors are classified according to their morphological and functional characteristics. Such neoplasms are identified as secreting or functioning when they produce hormones in sufficient amounts to lead to clinical manifestations, including, from high to low prevalence, lactotrophic tumors (prolactinomas), somatotropinomas (acromegaly, gigantism), corticotropinomas (Cushing’s disease), and, rarely, tumors secreting the glucoproteic hormones thyroid-stimulating hormone (TSH) (thyrotropinomas), luteinizing hormone (LH), and follicle-stimulating hormone (FSH) (gonadotropinomas). Pituitary adenomas can also secrete two or more hormones, with growth hormone (GH) and prolactin (PRL) co-secretion being the most prevalent of them. Pituitary neoplasms that do not produce circulating measurable amounts of intact hormones are called nonsecreting or nonfunctioning (NF) tumors.3 Regarding their morphology, pituitary tumors are classified as microadenomas, which are less than 10 mm in diameter, generally enclosed, and less frequently invasive, and macroadenomas, which expand the sellar boundaries and are invasive.4
 Clinical Presentation
Clinical Presentation
The clinical presentation can be broadly categorized based on (1) the tumor being incidentally found; (2) headaches due to compression or stretching of the dura lining of the sella or of the diaphragm, which is innervated by the branches of the trigeminal nerve; (3) mass effect on adjacent structures, such as the optic nerve and chiasm, or occasionally cranial nerves located in the cavernous sinus; and (4) endocrinologic dys-function. A rare emergent presentation, pituitary apoplexy, usually occurs with sudden-onset headache, meningismus, and oculomotor abnormalities, with a change in vision and disruption of the hypothalamic-pituitary-axis due to tumor hemorrhage or necrosis. However, pituitary hemorrhage occurs in up to 27% of cases of pituitary adenomas, and many of these have no clinical features of pituitary apoplexy.5
Optimal management of pituitary tumors requires a team approach. The unique location of the pituitary gland coupled with its complex role as the epicenter of control for hormone regulation can present unique obstacles for ideal treatment. For this reason, a series of examinations and investigations are recommended prior to surgery. These include neurologic, neuroendocrinologic, neuro-ophthalmologic, and neuroradiological assessments.
Pearl
• To obtain the optimal outcome, a team-based approach involving neurosurgery, ophthalmology, and endocrinology should be used for patient management.
 Endocrine Evaluation
Endocrine Evaluation
The endocrinologic screening is aimed at finding any hormonal deficits or hypersecretion. Pre- and postoperative endocrinologic screening includes evaluation of free cortisol, adrenocorticotropic hormone (ACTH), free thyroxine, TSH, PRL, GH, insulin-like growth factor-I (IGF-I), testosterone, estradiol, LH, and FSH to look for endocrinologic derangements. The diagnosis of a prolactinoma can be made based on serum PRL levels of > 150 ng/mL in combination with typical clinical symptoms.6 In patients with a prolactinoma, endocrinologic remission is defined as postoperative PRL levels of < 20 ng/mL in females or < 15 ng/mL in males. The diagnosis of Cushing’s disease is based on either abnormal 24-hour urinary free cortisol or abnormal results on low-dose dexamethasone suppression tests, defined as failure of 1 mg of dexamethasone to reduce plasma cortisol levels to < 1.8 mg/mL the next morning.7,8 The diagnosis of acromegaly is based on abnormal basal fasting levels of GH and IGF-I.9
Pearl
• A thorough history and physical examination should include questions about genetic syndromes such as multiple endocrine neoplasia (MEN) type 1. These individuals also have neoplasms of the parathyroid gland and the pancreas.
 Ophthalmologic Examination
Ophthalmologic Examination
A formal neuro ophthalmologic examination is essential in all patients. The examination includes visual field testing, both with confrontation and with Goldmann perimetry or semiautomatic perimetry. The classic bitemporal field loss is found in chiasmatic compression. Early compression may lead to upper quadrantic defects. This results from inferior chiasmal fiber compression. Evaluation of visual acuity with a Snellen chart without and with correction is essential. Funduscopy must be undertaken to evaluate the presence of optic nerve atrophy. Extraocular movements should be documented, especially in tumors extending into the surrounding cavernous sinus.
 Neuroimaging
Neuroimaging
Currently, magnetic resonance imaging (MRI) is the modality of choice for the diagnosis and characterization of a pituitary lesion. The standard protocol for MRI of the pituitary and parasellar region consists of sagittal T1- and T2-weighted images performed with and without intravenous contrast.10 Contrast enhancement may differentiate the adenoma from the displaced pituitary gland, may detect cavernous sinus invasion and demonstrate narrowing of the intracavernous internal carotid artery (ICA), and is helpful in the differential diagnosis of sellar and parasellar lesions. Thin coronal T2-weighted images provide good visualization of compression of the optic chiasm. Computed tomography (CT) is an alternative for patients with contraindications for MRI, and, in cases of bone invasiveness, CT bony window can be helpful in demonstrating the sellar floor and pneumatization of the sphenoid sinus.
A preoperative diagnosis of invasion is critical for planning surgical and adjuvant treatment strategies. Clinical signs of cavernous sinus invasion are evaluated on MRI. The Knosp- Steiner classification system uses this approach to preoperatively evaluate growth into the cavernous sinus (CS). Using mid-sella MRI in the coronal plane and the intra- and supra-cavernous segments of the carotid artery as reference points, a series of lines are drawn medially, through, and laterally to the ICA segments. The amount of tumor crossing these lines is divided into five grades (0 to 4). Grade 0 represents the normal condition with no invasion (the medial line is not crossed by tumor) and grade 4 represents total encasement of the ICA. Using 25 pituitary adenoma cases (functioning and nonfunctioning), Knosp et al11 correlated the preoperative classification with surgical confirmation of CS invasion, and it was determined that all grade 3 and 4 tumors exhibited CS invasion, most of those in the grade 2 category had surgically proven invasion, and the grade 0 and 1 had no invasion. The authors suggest that once the tumor crosses the intercarotid line (grade 2), CS invasion becomes very likely.
Pitfall
• Cavernous sinus invasion has classically been considered a feature of aggressive tumor behavior, and given the anatomic location, it is difficult to achieve gross total resection of tumors with CS invasion.
A number of authors have confirmed the usefulness of the Knosp classification in their own studies. In one study, it was used to show that none of the adenomas with parasellar extension grade 0 had CS invasion, and all adenomas with grade 4 had CS invasion when correlated with surgical outcome.12 There is also a correlation with Knosp criteria of CS invasion with surgical findings and MRI is also able to predict CS invasion with a sensitivity of 60% and a specificity of 85%.13
 Surgical Management
Surgical Management
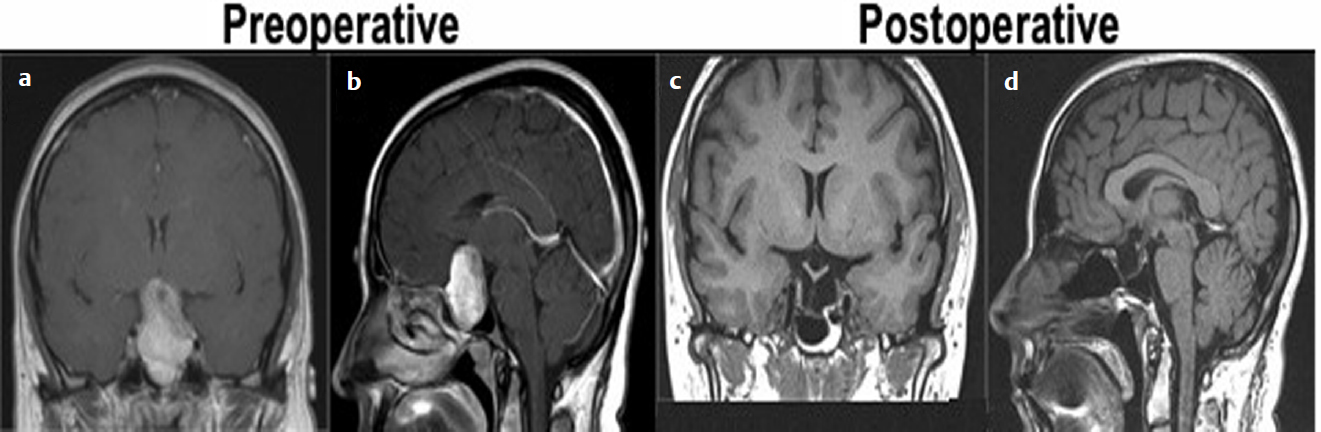
After appropriate medical workup and management, surgery usually provides the final diagnosis and indicates the appropriate treatment. Goals of surgery are (1) total tumor removal, (2) decompression of the optic chiasm and nerve, (3) tumor debulking for cytoreduction, (4) preservation or restoration of endocrine function, and (5) histological confirmation. The last few decades have been marked by advances in diagnostic and surgical instruments and technology. They provide the neurosurgeon with a large armamentarium to help achieve surgical goals while reducing complications. They include, but are not limited to, intraoperative imaging (intraoperative CT or MRI14), neuronavigation, handheld Doppler ultrasound for carotid artery identification, use of indocyanine green to differentiate tumor margins,15 and endoscopic endonasal instrumentation (two- and three-dimensional endoscopes). Controversies over ideal surgical approach (craniotomy versus microscopic endonasal versus endoscopic endonasal) have declined as larger surgical series have been reported with longer follow-up. Neurosurgeons in training and in practice are becoming more comfortable with the evolving technology (Fig. 34.1).
The advantages of an endonasal method over a craniotomy include a less invasive approach, a more direct anatomic route, no craniotomy or facial incisions, less trauma to the brain and neurovascular structures, early devascularization of the tumor blood supply, improved visualization of relevant anatomy, a better cosmetic result, and shorter recovery times. The endoscope also provides a wider field of view compared with the microscope. With advances of sellar floor reconstruction, for example with a vascularized nasoseptal flap,16 the problem of greater cerebrospinal fluid leaks has become less prominent for surgery on adenomas isolated to the sella.
 Individual Pituitary Tumor Types
Individual Pituitary Tumor Types
Prolactinomas
Hyperprolactinemia is among the most common of pituitary disorders, and prolactinomas account for 30 to 60% of pituitary tumors.17 Physiological hyperprolactinemia is seen with physical and emotional stress, pregnancy, nipple stimulation, and after sexual orgasm. Iatrogenic elevation occurs by antagonizing dopamine action with such medications such as antiemetics, antidepressants, antipsychotics, and narcotics. The clinical findings, regardless of the patient’s sex, can be associated with anxiety, depression, fatigue, emotional instability, and hostility.18,19 Symptoms in women of reproductive age include amenorrhea, galactorrhea, infertility, seborrhea, and hirsutism. Low estrogen can result in a loss of libido, and long-lasting effects include osteopenia. In men, the most common presentation is a loss of libido and impotency, and less commonly oligospermia and hypogonadism. Galactorrhea or gynecomastia is present in 15 to 30% of male patients.20
Treatment goals are dependent on acuity and cause of presentation, with the ultimate goal being normalization of prolactin levels. Patients presenting with noniatrogenic hyperprolactinemia are usually treated medically with dopamine agonists to normalize serum levels but also to control tumor size and growth. A systematic review of the use of cabergoline and bromocriptine for prolactinomas found that cabergoline was more effective at normalization of hyper-prolactinemia and associated with significantly less adverse events.21 Patients requiring surgery have either failed medical treatment or are experiencing major adverse effects induced by all of the dopaminergic agonists. There is still some controversy as to the use of cabergoline, given the long-term adverse effects when a number of surgical series report remission rates of 85 to 89%22–24 with recurrence rates of 18.7%.25
Pitfall
• In giant prolactinomas the “hook effect” is described as a low serum prolactin level, when in reality if the samples are diluted (10-fold dilution) there is a substantially high level.
Acromegaly
Acromegaly is a disease of chronic overproduction of GH. The consequences of GH oversecretion are numerous and include, but are not limited to, facial changes (large lips, tongue, skin changes), laryngeal hypertrophy (low voice), bony hyper-trophy (prognathism, thick skull, jaw, hands, cervical spine stenosis), hypertension, cardiomyopathy, barrel chest, high adrenocorticoid output, and chronic renal volume increase. Acromegaly is diagnosed by clinical features, an elevated serum IGF-I level, and a serum GH level that does not decline to < 1 ng/mL after oral glucose (75 or 100 g). The definitive test for acromegaly is the GH response to an oral glucose challenge (oral glucose tolerance test [OGTT]). The test must be performed correctly to interpret the results. Baseline serum glucose and GH are measured, then the patient drinks a glucose solution (75 or 100 g), and the serum glucose and GH levels are measured every 30 minutes for 2 hours. The current guideline for a normal response is a serum GH level of < 1 ng/mL. Cardiac disease is the most important cause of morbidity and mortality in acromegalic patients.26,27 This is followed by respiratory disease where upper airway obstruction (obstructive sleep apnea) affects up to 70% patients.28
Surgery remains the first-line therapy.9 Whether microscopic or endoscopic, the surgical techniques are the same as with other adenomas. Remission rates vary between 46% and 85%: for microadenomas, 75 to 100%; for macroadenomas, 50 to 80%.29–32 Intraoperative biochemical testing to determine remission during resection has been employed in some centers, with successful measurement of intraoperative GH levels as a guide to remission.33
• The relative importance of GH and IGF-I values in the postoperative follow-up is not clear. Optimal levels of both appear to be required to maintain normal health.
Cushing’s Disease
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


