Fig. 1
Histological section of normal thyroid, magnified 20 ×. Each colloid-filled follicle is surrounded by a single layer of thyroid follicular cells
Thyroid Hormone Synthesis
The thyroid gland produces two main types of TH: thyroxine (T4) and triiodothyronine (T3). They share a nearly identical molecular structure—phenolic rings joined by an ether link and iodinated at either three (T3) or four (T4) positions (Fig. 2). Since only T3 is biologically active, T4 is considered a prohormone and requires conversion to T3 in the peripheral tissues [2]. Both types of TH are synthesized in the follicular cells, a process that involves concentration of iodine, organification and coupling, and, finally, secretion or storage [2, 4] (Fig. 3).
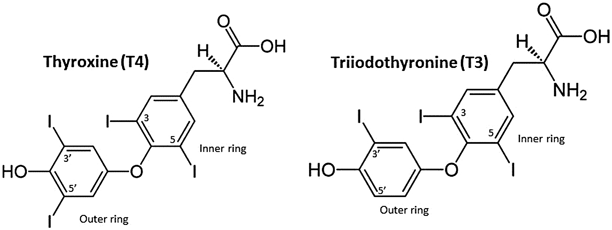
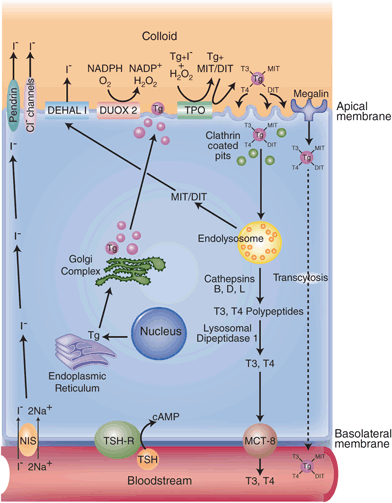
Fig. 2
The molecular structures of thyroxine ( T4) and triiodothyronine ( T3)
Fig. 3
The synthesis and secretion of thyroid hormone by the thyroid follicular cell. Thyroid hormone synthesis is stimulated by binding of TSH to the TSHR, which increases intracellular cAMP. Iodide enters the thyroid gland via the Na+/I- symporter ( NIS). The intracellular electrochemical gradient then drives I- to the apical surface of the cell, where it exits into the follicular lumen either through pendrin or chloride channels or both. The I- is then oxidized by the enzyme thyroperoxidase ( TPO) in the presence of hydrogen peroxide (H2O2), which is generated by dual oxidase 2 (Duox2), an NADPH oxidase. Oxidized iodide attaches to tyrosyl residues of thyroglobulin to form monoiodotyrosine ( MIT) or diiodotyrosine ( DIT) residues, a process also catalyzed by TPO. MIT and DIT then combine to form T3 and T4, all still incorporated within the thyroglobulin molecule. Iodinated thyroglobulin is then taken up by clathrin-coated pits on the apical surface of the follicular cell, which invaginate to form intracellular vesicles. These fuse with the endolysosomal system. MIT, DIT, T4, and T3 residues are selectively removed from the molecule. MIT and DIT residues are deiodinated by DEHAL1 and the iodide is recycled. The T3- and T4-containing polypeptides are further processed to become free T3 and T4, which are then released into the circulation via a hormone transporter, MCT8. A small proportion of iodinated thyroglobulin binds to a protein called megalin before being internalized by the thyrocyte and is transported through the cell before being released intact into the bloodstream. This process is called transcytosis. TSH thyroid-stimulating hormone, TSHR thyroid-stimulating hormone receptor, NADPH nicotinamide adenine dinucleotide phosphate, MCT monocarboxylate transporter
Iodine Metabolism
Only 7 years after the discovery of iodine in 1812, Coindet published a description of the element being used for the treatment of goiter [6, 7]. Throughout the nineteenth century, observational evidence mounted connecting iodine deficiency to goiter, cretinism, and myxedema, but the presence of iodine in the thyroid gland was not formally demonstrated until 1896, and TH was not isolated until 1919—exactly 100 years after Coindet’s publication [7] .
Iodine’s role as a key ingredient for TH synthesis (it accounts for 65 % of the molecular weight of T4 and 59 % of T3) is now well understood [8]. Iodine is an essential micronutrient that can be acquired only via dietary sources [6, 8, 9]. It circulates in the bloodstream in its ionized form, iodide (I-). Since up to 90 % of ingested iodine is excreted in the urine, the thyroid evolved a very efficient mechanism to “trap” and concentrate iodide [6, 10, 11]. In order to provide the adult thyroid with the 60 µg of I- per day required to synthesize normal amounts of TH, the recommended daily intake of iodine is 150 µg [6, 9, 12]. Under normal physiologic conditions, the thyroid contains more than 90 % of the total body iodide, with an I- concentration 20–40 times higher than that of serum [13].
The mechanism for the gland’s ability to accumulate I- against its concentration gradient was a subject of considerable research and debate until the cloning of the Na+/I- symporter (NIS) in 1996 [6, 14]. Located at the basolateral plasma membrane of the follicular cells, the NIS has 13 transmembrane segments [6, 15]. It uses the inwardly directed Na+ gradient generated by the Na+/K+ ATPase to couple movement of Na+ and I- from the blood to the follicular cell cytoplasm. Two Na+ ions enter for every I-, and this 2:1 stoichiometry generates a positively charged inward current [6, 15]. Activity of the NIS and uptake of iodide are regulated by thyroid-stimulating hormone (TSH) and by iodide itself [15] .
Once I- is inside the follicular cell, the electrochemical gradient drives it to the apical surface of the cell. The mechanism of the efflux of I- from the cell cytoplasm into the follicular lumen is less well defined [13, 16]. Another transmembrane protein, pendrin, is thought to play a role in apical iodide transport. This anion exchanger is mutated in Pendred’s syndrome, leading to a partial defect in organification of iodide. The syndrome is characterized by hereditary sensorineural deafness and occasionally euthyroid goiter [6, 13, 15, 16]. However, the fact that Pendred’s syndrome patients living in iodine-replete regions do not tend to develop goiter, combined with the findings that pendrin knockout mice have normal thyroid size and function, suggests that other routes for I- efflux exist as well [6, 13, 16]. Several anion transporters and channels known to transport chloride, including the Cl-/H+ antiporter (CLC-5), the sodium-monocarboxylic acid transporter (SMCT1), and the cystic fibrosis transmembrane conductance regulator (CFTR), have been proposed as alternate candidates to mediate the translocation of I- to the follicular lumen [6, 16] .
Before it can be incorporated into the tyrosine residues on thyroglobulin, I- must be oxidized [2, 13]. This process is catalyzed by the enzyme thyroperoxidase (TPO) and requires the presence of hydrogen peroxide (H2O2) [8, 13]. A multispanning membrane protein called dual oxidase 2 (Duox2), a calcium-dependent nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, plays a key role in generating the necessary H2O2 on the luminal surface of the apical follicular cell membrane [6, 13]. Although the substrates and catalyst for this reaction are well understood, the exact nature of the oxidized iodinating species is still not known [17, 18]. Several models have been proposed, including hypoiodite (OI-), hypoiodous acid (HOI), or iodonium (I+) [13, 18] .
Thyroglobulin
Synthesized in the follicular cells, thyroglobulin is the most highly expressed protein in the thyroid gland [19]. With a dimeric molecular mass of 660 kDa, it is one of the largest proteins in the human body, and it consists of a signal peptide sequence and approximately 2750 amino acid residues [19, 20]. The protein has a homogeneous structure in all vertebrates, consisting of four distinct regions: three cysteine-rich repetitive domains (Tg1, Tg2, and Tg3) and the cholinesterase-like (ChEL) domain (C-terminus of the molecule), which is highly homologous to acetylcholine [19, 21, 22]. Production of thyroglobulin is regulated by TSH and several transcription factors, including thyroid transcription factor 1 (TTF-1), TTF-2, and paired box gene 8 (Pax-8) [19]. As the nascent thyroglobulin polypeptide chains emerge from the polyribosomes, they enter the cytosolic side of the endoplasmic reticulum, where each region undergoes an independent oxidative folding process [19, 22, 23]. All the cysteine residues ultimately become incorporated into disulfide bonds, although the number of intra/interchain bonds creates significant potential for errant folding [22, 23]. Thyroglobulin molecules also undergo glycosylation and dimerization [19, 21]. Several molecular chaperones, including BiP, GRP94, GRP170, ERp29, ERp72, and calnexin, are involved in the maturation of thyroglobulin and act as intracellular quality control to ensure that only properly folded and glycosylated thyroglobulin molecules are ultimately released [8, 19, 20]. The process is relatively lengthy, with a half-time of 90–120 min for newly synthesized thyroglobulin to arrive at the Golgi complex [22]. There it is incorporated into exocytic vesicles that migrate to the apical membrane of the follicular cell, where the organification and coupling process takes place [8, 19].
Organification and Coupling
Although organification and coupling are often discussed as discrete steps, both reactions are catalyzed by TPO and in fact occur simultaneously [13]. The process by which oxidized iodide attaches to tyrosyl residues of thyroglobulin is referred to as organification. The reaction leads to the formation of monoiodotyrosine (MIT) or diiodotyrosine (DIT) residues, still located within the thyroglobulin molecule [8, 13]. Of the 132–140 tyrosyl residues in each molecule of dimerized human thyroglobulin (the reported number varies because of refinements in complementary DNA (cDNA) sequencing), only a small subset are actually susceptible to iodination [8, 13, 21, 24]. While each molecule of mature thyroglobulin contains 5–50 iodine atoms, only certain iodinated sites are ultimately involved in hormonogenesis because of their spatial orientation [8, 13].
Once MIT and DIT form, the coupling reaction occurs: two DIT residues combine to form T4 or one MIT combines with one DIT to create T3 [8, 13]. In this reaction, also catalyzed by TPO and mediated by H2O2, the iodinated phenyl group of a tyrosyl residue is donated to become the outer ring of the iodothyronine acceptor site [13]. Tyrosines 5, 2554, and 2747 are the most important acceptor sites, while tyrosines 130, 847, and 1448 are the dominant donors [13, 24]. About one-third of T4 is created by the acceptor–donor pair of tyrosine 5 and tyrosine 130 [8]. After the coupling reaction, the newly formed T3 and T4 are still incorporated within the thyroglobulin molecule [8]. Under normal conditions (normal iodine supply and normal thyroid function), the average thyroglobulin molecule contains 2.5 residues of T4 residues, 0.7 of T3, 4.5 of DIT, and 5 of MIT [8].
Secretion and Storage
Once organification and coupling are complete, the mature, iodinated thyroglobulin is released into the follicular lumen, where it comprises more than 95 % of the colloid [8, 25]. About two-thirds of thyroglobulin in the colloid is in the soluble 660 kDa dimerized form, but free monomers and tetramers are also found in minimal amounts [8, 25]. The other 34 % of thyroglobulin exists in a dense, highly concentrated insoluble form (i-Tg), with high iodine content but nearly undetectable TH residues [8, 11, 25].
When follicular cells are stimulated by TSH, thyroglobulin is mobilized from the colloid and reenters the thyrocyte via a process called micropinocytosis. Because of their physical proximity to the apical membrane and solubility, newly secreted thyroglobulin molecules are taken up first (the “last-come-first-served” principle first described by Schneider et al. in 1964) [11, 25, 26]. Clathrin-coated pits form on the luminal side of the apical membrane of thyrocytes and internalize thyroglobulin. These pits then invaginate to form intracellular vesicles, which shed their clathrin coats and quickly fuse with early endosomes [8, 25, 27]. This process is thought to occur via a combination of nonspecific fluid-phase uptake (which likely is the main route of thyroglobulin uptake leading to degradation and TH release) and receptor-mediated uptake (which appears to lead to other post-endocytic pathways) [8, 25, 28].
The intracellular thyroglobulin molecules then follow one of at least three different pathways: proteolytic destruction into its component parts, recycling back to the follicular lumen, or transcytosis [8, 25, 28]. In the first pathway, thyroglobulin travels through the endosomal system to lysosomes, where MIT, DIT, T4, and T3 residues are selectively removed from the molecule [8, 29]. Free MIT and DIT residues released from lysosomes cannot combine to synthesize hormone; rather, they are deiodinated to produce I- and tyrosine, both of which are then reused for novel thyroglobulin synthesis and organification . The enzyme primarily responsible for this iodide recycling process is iodotyrosine dehalogenase 1 (DEHAL 1), an NADPH-dependent transmembrane protein located at the apical membrane of follicular cells [30, 31]. Finally, cathepsins D, B, and L, which are lysosomal proteases, act at specific cleavage sites on thyroglobulin to produce hormone-enriched polypeptides. These are then further hydrolyzed by exopeptidases (most importantly lysosomal dipeptidase I) to release free TH into the cytoplasm [32–34].
Conventionally, the secretion of TH into the bloodstream has been attributed to passive diffusion, but more recent evidence supports a transport system located at the follicular cell basolateral membrane. Over the past few decades, transmembrane TH transporters, most notably monocarboxylate transporter 8 (MCT8) and OATP1C1 (a member of the sodium-independent organic anion-transporting polypeptide family), have been well described in peripheral tissue. Recent animal studies have demonstrated immunohistochemical localization of MCT8 to the basolateral membrane of the thyrocyte and have also suggested that MCT8 is in fact necessary for normal TH secretion from the thyroid gland [35, 36].
Although the most important post-endocytic pathway for thyroglobulin involves proteolysis and production of free hormone, not all thyroglobulin taken up by follicular cells is processed in the lysosomes. Immature thyroglobulin molecules tend to be iodine poor and incompletely glycosylated [8]. Several receptors, including the thyroid asialoglycoprotein receptor and the putative N-acetylglucosamine receptor, as well as the molecular chaperone protein disulfide isomerase (PDI), have been proposed to recognize these immature characteristics and facilitate the recycling of immature thyroglobulin back to the follicular lumen, but their exact roles remain unclear [8, 25]. Megalin, another receptor found on the luminal side of the follicular cell’s apical membrane, is known to mediate the third pathway for internalized thyroglobulin called transcytosis. Thyroglobulin that binds to megalin before being internalized by the thyrocyte escapes the lysosomal pathway and instead is transported intact to the basolateral membrane with subsequent release into the bloodstream [37, 38]. With normal TSH stimulation, the expression of megalin is relatively low and very little thyroglobulin is transcytosed. When TSH is elevated, however, megalin expression increases, leading to significantly greater amounts of transcytosis. By diverting thyroglobulin from lysosomal proteolysis, this may serve to reduce free hormone release under conditions of massive TSH stimulation [25, 38].
Peripheral Actions of Thyroid Hormone
Under normal circumstances, 90 % of the hormone secreted by the thyroid gland is in the form of T4 and the other 10 % is T3 [2]. Only a very small fraction of the released TH (about 0.03 % of total T4 and 0.3 % of total T3) circulates as free hormone [39]. The rest of the released TH binds to carrier proteins, with 95 % bound to either thyroid-binding globulin (TBG), transthyretin (TTR), or albumin. The remaining 4–5 % is bound to other minor TH carriers, including lipoproteins, immunoglobulins, and lipocalins [39, 40]. These carrier proteins serve both as a stable reservoir of extrathyroidal TH and as protection for the extremely hydrophobic free hormone from its aqueous environment [2, 39, 40]. Because of this method of distribution and buffering, T4 has the longest half-life of any hormone (6–7 days), and its serum concentration of 0.1 µM is second only to cortisol [40].
Circulating free TH can be taken up by peripheral cells at any time, and both T4 and T3 can rapidly dissociate from their carrier proteins to increase the availability of free hormone when necessary [2, 40]. The entry of free T4 or T3 into target cells is mediated by a variety of plasma membrane transporters, including the aforementioned monocarboxylate transporters (including MCT8 and MCT10) and OATP1C1, as well as the Na+ -taurocholate cotransporting polypeptide, the L-type amino acid transporters (LAT1 and LAT2), and fatty acid translocase [35, 36, 41]. To date, the roles of the MCT and OATP families are best understood. While the MCT transporters are widely expressed in most tissues, OATP1C1 is much more specific, expressed predominantly in the brain and testes [36, 42]. The transporters also vary in their ligand specificity, with MCT10 demonstrating increased affinity for T3 and OATP1C1 demonstrating strong preference for T4 [36, 42]. The exact mechanisms by which these transporters facilitate TH uptake are still under investigation [35, 41, 42]. The clinical importance of TH transporters is manifested in the Allan–Herndon–Dudley syndrome. First described in 2004, this X-linked syndrome of severe psychomotor retardation and elevated T3 levels, results from mutations in the MCT8 gene [41, 42].
Once inside the cytosol of the target cells, T4 (and sometimes T3) is deiodinated by the iodothyronine deiodinase family of selenoproteins. Outer (5¢)-ring deiodination of the prohormone T4 produces the active form of T3, while inner (5)-ring deiodination produces the inactive metabolite (reverse T3; rT3) [43, 44]. Although they share an overall 50 % sequence homology and similar catalytic properties, the three deiodinases differ with regard to function and tissue specificity [2, 43, 45].
Type 1 deiodinase (D1) , anchored on the cytosolic side of the plasma membrane, is the only enzyme of the three that is capable of deiodinating either the inner or the outer ring [43, 44]. Although it is believed to be an important source of T3 to the circulation, D1 is much less efficient than D2 in catalyzing the T4 to T3 conversion [44]. D1 also displays a strong preference for rT3 as its outer ring deiodination substrate. Consequently, it has been proposed that the more important role of D1 is as a scavenger enzyme, clearing inactive iodothyronines from the circulation and recycling the iodine [43, 44]. It is primarily expressed in the thyroid, liver, and kidney [43]. Increased thyroidal activity of D1 is thought to play a major role in the disproportionate overproduction of T3 observed in Graves’ disease and multinodular toxic goiter, while reduced hepatic and renal D1 activity may account for the decreased levels of T3 associated with nonthyroidal illness syndrome (NTIS) [44].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


