The evolution of understanding cancer biology has yielded many advances that have been translated into cancer treatment. Application of this knowledge has allowed for a shift in chemotherapeutics from traditional cytotoxic agents that worked by killing both healthy and malignant fast growing cells to chemical and biologic therapies aimed at targeting a specific gene or pathway critical to the particular cancer being treated.1 This age of pathway-directed therapy has been made possible by the increased availability and feasibility of high throughput technology able to provide comprehensive and clinically useful molecular characterization of tumors. Translation of these efforts have resulted in improved degree to disease control for many common cancers including breast, colorectal, lung, and melanoma as well as long-term survival benefits for chronic myelogenous leukemia (CML), gastrointestinal stromal tumors (GIST), and childhood acute lymphoblastic leukemia (ALL).2
Pharmacogenomic-guided therapy aims the use information on DNA and RNA integrity to optimize not only the treatment choice for an individual patient, but also the dose and schedule of that treatment. The assessment of both somatic and germ-line mutations contribute to the overall individualization of cancer treatment. Somatic mutations are genetic variations found within the tumor DNA, but not DNA from the normal (germ-line) tissues, which also have functional consequences that influence disease outcomes and/or response to certain therapies. These types of mutations or biomarkers can be classified as either prognostic or predictive. Prognostic biomarkers identify subpopulations of patients with different disease courses or outcomes, independent of treatment. Predictive biomarkers identify subpopulations of patients most likely to have a response to a given therapy.3 Germline mutations are heritable variations found within the individual and, in practical terms, are focused on DNA markers predictive for toxicity or therapeutic outcomes of a particular therapy as well as inheritable risk of certain cancers.4 Pharmacogenomic mutations in the germ line provide some explanation for the interindividual and interracial variability in drug response and toxicity. For cancer chemotherapy, where cytotoxic agents are administered at doses close to their maximal tolerable dose, and therapeutic windows are relatively narrow, minor differences in individual drug handling may lead to severe toxicities. Therefore, an understanding of the sources of this variability would lead to the possibility of individualizing dosages or influencing clinical decisions that can improve patient care. Pharmacogenomics has putative utility in therapy selection, clinical study design, and as a tool to improve understanding of the pharmacology of a medication.
The term pharmacogenetics was initially used to define inherited differences in drug effects and typically focused on individual candidate genes. The field of pharmacogenomics now includes genomewide association studies and is used to describe genetic variations in all aspects of drug absorption, distribution, metabolism, and excretion in addition to drug targets and their downstream pathways.5Table 16.1 illustrates some current clinical examples of genotype-guided cancer chemotherapy. Variations in the DNA sequences encoding these proteins may take the form of deletions, insertions, repeats, frameshift mutations, nonsense mutations, and missense mutations, resulting in an inactive, truncated, unstable, or otherwise dysfunctional protein. The most common change involves single nucleotide substitutions, called single-nucleotide polymorphisms (SNP), which occur at approximately 1 per 1,000 base pairs on the human genome. Variability in toxicity or activity can also be mediated by postgenomic events, at the level of RNA, protein, or functional activity.
PHARMACOGENOMICS OF TUMOR RESPONSE
Tumor response to chemotherapy is regulated by a complex, multigenic network of genes that encompasses inherent characteristics of the tumor, differentially activated pathways of cell signaling, proliferation and DNA repair, factors that control drug delivery to the tumor cells (e.g., metabolism, transport), and cell death. These may in turn be modulated by previously administered treatment or drug exposure, which may upregulate target proteins or activate alternative pathways of drug resistance. The polygenic nature of drug response implies that a better understanding of genotype-phenotype associations would require more than the usual single-gene pharmacogenetic strategies employed to date. However, there are instances where the genomic context of a single gene within a cancer will be of high impact for specific therapeutic agents (see Table 16.1).
Pathway Directed Anticancer Therapy
One of the earliest success stories illustrating pathway-driven therapeutics is with CML. The hallmark chromosomal abnormality of this disease is the translocation of chromosomes 9 and 22 that ultimately produces the fusion gene BCR-ABL. This discovery in 1960 eventually led to the development of the targeted tyrosine-kinase inhibitor (TKI) imatinib and its subsequent Food and Drug Administration (FDA) approval for treatment of CML in 2001.6 The International Randomized Study of Interferon and STI571 (IRIS) trial began enrollment in 2000 and compared imatinib with interferon and low-dose cytarabine, which was the previous standard of care for newly diagnosed patients with chronic-phase CML. All efficacy endpoints favored imatinib, including complete cytogenetic response of 76.2% with imatinib compared with 14.5% with interferon (p <0.001).7 Overall survival (OS) after 60 months of follow-up was 89% with imatinib.8 This example is just one of many where a once fatal disease can now be considered more akin to a chronic disease, requiring a daily medication and regular physician follow-up, similar to hypertension or diabetes. Drug development has also kept pace with these advances and now several other agents, including dasatinib, nilotinib, bosutinib, and ponatinib, have joined imatinib as treatment options for CML.
The idea of changing treatment focus from a disease-based model to a pathway-driven model is also evolving. Human epidermal growth factor receptor 2 (HER2) is a transmembrane receptor tyrosine kinase that is overexpressed or amplified in up to 25% of breast cancers. Trastuzumab is a humanized monoclonal antibody directed against HER2 and demonstrated improved response rates (RR) and time to disease progression in patients with metastatic HER2 positive breast cancer and improved disease-free survival (DFS) and OS in HER2-positive breast cancer patients treated with adjuvant trastuzumab.9 Several additional agents are now available to target the HER2 pathway and vary in their pharmacology and mechanism of action. Lapatinib is an oral TKI directed against HER2 and the epidermal growth factor receptor (EGFR), pertuzumab is a humanized monoclonal antibody that binds at a different location than trastuzumab and inhibits the dimerization and subsequent activation of HER2 signaling, and ado-trastuzumab emtansine is an antibody-drug conjugate that targets HER2-positive cells and then releases the cytotoxic antimitotic agent emtansine through liposomal degradation of the linking compound. All of these agents illustrate the progress and pharmacologic diversity of pathway-directed therapy and remain as standard of care options for HER2-positive breast cancer in either the adjuvant and/or metastatic settings.10 HER2 expression is not limited to breast cancer, however. Though less common, HER2 expression is seen in numerous solid tumors including bladder, gastric, prostate and non-small-cell lung cancer with varying degrees of incidence depending on the method of detection. Based on results from a large, open-label phase III randomized, international trial of 594 patients with gastric or gastroesophageal junction cancer expressing HER2 by either immunohistochemistry or gene amplification by fluorescence in situ hybridization, trastuzumab is also approved for treatment of metastatic gastric or gastroesophageal junction adenocarcinoma that expresses HER2. Patients randomized to chemotherapy in combination with trastuzumab had a median OS of 13.8 months compared with 11.1 months in the patients receiving chemotherapy alone (hazard ratio [HR], 0.74; 0.60 to 0.91, p = 0.0046).11 Numerous examples also support that pathway-directed therapy will cross the boundaries of disease sites and that tumor genetics will become one of the biggest determining factors for treatment.
TABLE 16.1 Clinical Examples of Genotype-Guided Cancer Chemotherapy
Decreased serum levels of active drug and potential decreased efficacy in patients with high enzyme levels (ultrarapid metabolizers)
CYP2D6
Tamoxifen, codeine, ondansetron
Decreased production of active metabolite and potential decreased efficacy in patients with low enzyme levels
Dihydropyrimidine dehydrogenase (DPYD)
5-Fluorouracil
Decreased elimination and increased risk of myelosuppression, diarrhea, and mucositis in patients with low enzyme levels
Glucose-6-phosphate dehydrogenase (G6PD)
Rasburicase
Risk of severe hemolysis in patients with G6PD deficiency
Thiopurine methyltransferase (TPMT)
Mercaptopurine, thioguanine, azathioprine
Decreased methylation of the active metabolite resulting decreased elimination and increased risk of neutropenia in patients with low enzyme levels
UDP-glucuronosyltransferase (UGT) 1A1
Irinotecan
Decreased glucuronidation of the active metabolite resulting decreased elimination and increased risk of neutropenia and diarrhea in patients with low enzyme levels
Simple expression of the drug target does not always translate into desired clinical outcomes though. Cetuximab and panitumumab are monoclonal antibodies directed against EGFR; however, it was found that colorectal cancer (CRC) patients who did not have detectable EGFR still experienced responses to these agents similar in extent to EGFR-positive patients. Kirsten rat sarcoma viral oncogene (KRAS) is a downstream effector of the EGFR pathway. Ligand binding to EGFR on the cell surface activates pathway signaling through the KRAS-RAF-mitogen-activated protein kinase (MAPK) pathway, which is thought to control cell growth, differentiation, and apoptosis.12 Eventually it was found that CRC patients with a KRAS mutation did not derive benefit from cetuximab or panitumumab. The RR in CRC receiving either cetuximab or panitumumab who were KRAS wild type was 10% to 40% compared with near zero percent in those with KRAS mutations.13 This finding was the result of a retrospective analysis of small group of patients and was confirmed in large, prospective trials. Additionally, it underscores the importance of tissue collection for biomarker assessment in trials with novel therapeutics. A recent clinical trial genomic analysis suggests that mutations in NRAS may also have value in predicting the utility of EGFR antibody therapy in colorectal cancer. Although the predictive value of KRAS mutation status in colorectal cancer has been well established in clinical trials, the role of KRAS in lung cancer and other malignancies is less well elucidated. Lung cancers harboring KRAS mutations have been shown to have less clinical benefit from the EGFR-targeted erlotinib in some trials, although this has not consistently been the case across all trials. Additionally, lung cancer KRAS mutation status does not appear to reproducibly predict clinical benefit from the EGFR-targeted monoclonal antibodies, as is the case in colorectal cancer.14 Unlike the HER2 example discussed previously, the clinical application of some genetic mutations will differ between tissue of origin.
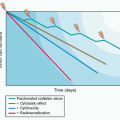
Deeper investigations and understandings of mutations driving oncogenic pathways can also elucidate mechanisms of resistance and practical therapeutic strategies for treatment and prevention. Approximately half of all cutaneous melanomas carry mutations in BRAF, with the most common being the V600E mutation. Vemurafenib is a TKI directed against mutated BRAF that demonstrated improvements in both progression-free survival (PFS) and OS when compared with the cytotoxic agent dacarbazine in previously untreated patients with metastatic melanoma carrying the BRAF V600E mutation. Vemurafenib demonstrated a 63% relative reduction in the risk of death compared with dacarbazine (p <0.001) along with a higher response rate (48% compared with 5% for dacarbazine).15 Based on these results, vemurafenib was the first BRAF targeted TKI approved by the FDA and was soon joined by dabrafenib. Although dramatic responses to these agents have been observed, relapse almost universally occurs after a median of 6 to 8 months. Activating BRAF mutations, like V600E, result in uncontrolled activity of the MAPK pathway through activation of the downstream kinase MEK, which when phosphorylated, subsequently activates extracellular signal-regulated kinase (ERK), which ultimately translocates to the cell nucleus, resulting in cell proliferation and survival (Fig. 16.1).16 An assessment of serial biopsies from patients treated with vemurafenib suggested numerous mechanisms for acquired resistance, including the appearance of secondary mutations in MEK.17 This finding supports the clinical rationale for using combination therapy with a BRAF and a MEK inhibitor. The combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) was assessed in 247 metastatic melanoma patients with BRAF V600 mutations compared with dabrafenib alone. Median PFS was 9.4 months in the combination group compared with 5.8 months in the patients who received single agent therapy (HR, 0.39; 0.25 to 0.62, p <0.001). A complete or partial response was also higher in the combination therapy group (76% compared with 54%, p = 0.03). The occurrence of cutaneous squamous cell carcinoma, a known side effect of single-agent BRAF inhibitor therapy due to paradoxical activation of RAF in nonmutated cells, was also decreased in the combination therapy group (7% compared with 19%, p = 0.09), further supporting the evidence of downstream inhibition.18 Although combination therapy does prolong the time to disease progression, resistance still occurs in patients through a variety of mechanisms. Utilization of sequential biopsies and a genetic assessment will help to inform rationale combination and sequential pathway-driven therapy trials that will ultimately aid in better understanding and mitigation of common mechanism of resistance.
Only gold members can continue reading. Log In or Register to continue