29 Pediatric Supratentorial Tumors
Tumors of the central nervous system (CNS), as a group, make up approximately 25% of all childhood cancers and represent the most common solid tumors in children. Supratentorial and infratentorial tumors occur with almost equal incidence in the pediatric population. However, the relative frequency by location varies according to the age of the patient, with supratentorial tumors predominating in children up to age 3 and older than age 10.1 Glial-based tumors make up 60% of neoplasms within the supratentorial compartment, and 80% of these are of low-grade histology.2 This chapter reviews several supratentorial neoplasms in children. Some important supratentorial pediatric tumors, such as craniopharyngiomas and pineal region tumors, are discussed in other chapters.
 Clinical Presentation in Childhood
Clinical Presentation in Childhood
Early diagnosis of CNS tumors in the pediatric population is a primary determinant of treatment and outcome. The symptoms of initial presentation generally relate to the tumor location and the rate of growth. For instance, tumors within the frontal lobe may lead to personality changes, seizures, or headaches. A tumor located in the temporal lobe may cause seizures or speech alterations. Tumors of the suprasellar region typically present with endocrinopathies or visual changes. Those within the thalamus usually result in motor and sensory deficits. Tumors of the tectal plate and pineal region may lead to obstructive hydrocephalus, as do tumors invading or emanating from the lateral and third ventricles. However, some symptoms may be nonlocalizing, such as vomiting. Often misdiagnosed as a gastrointestinal disease or migraine variant, it is one of the most common presentations of childhood CNS tumors.
Pearls
• Primary CNS tumors are more common in Caucasians than in African Americans (5.02 vs 3.69 per 100,000) and more common in males than in females (4.9 vs 4.8 per 100,000).
• Two factors are reliably associated with an increased risk of developing a primary CNS tumor in childhood: a history of significant doses of radiation to the CNS, and the diagnosis of certain genetic syndromes (neurofibromatosis, Li-Fraumeni syndrome, tuberous sclerosis, von Hippel–Lindau syndrome, Gorlin syndrome, Turcot syndrome, Cowden syndrome, etc.).
The confirmation of a suspected brain tumor ultimately relies on some type of neuroimaging. The decision of whether to obtain a computed tomography (CT) or magnetic resonance imaging (MRI) examination of the brain with an acute presentation in the emergency room is usually relatively straightforward. In contrast, primary care pediatricians may be following more subtle signs on a chronic basis and face a more challenging decision about whether imaging is justified.
 High-Grade Tumors
High-Grade Tumors
High-Grade Astrocytomas
Anaplastic astrocytomas are classified as World Health Organization (WHO) grade III lesions and are characterized by rapid and infiltrative growth. They tend to occur at about the same frequency in children as lower grade diffuse astrocytomas.3 When supratentorial, these tumors predominantly occur in the cerebral hemispheres, but may also be found in deep midline structures. Microscopically, anaplastic astrocytomas are diffusely infiltrating, with increased cellularity, nuclear pleomorphism, and high mitotic activity. Chromosomal gains of 5q and losses of 6q, 9q, 12q, and 22q are characteristic aberrations in anaplastic astrocytomas in children. Furthermore, significantly shorter survival has been noted in patients whose tumors display gain of material at chromosome arm 1q.4
Glioblastoma multiforme (WHO grade IV) occurs approximately 1.5 times more often than anaplastic astrocytoma, but is about 100 times less common in children than in adults. Combined frontotemporal location is typical, but parietal and occipital lobes may also be affected. Glioblastomas are diffusely infiltrating tumors defined by the presence of prominent microvascular proliferation or necrosis. These highly malignant tumors typically demonstrate high cellularity with marked nuclear atypia and brisk mitotic activity.
Genomic amplification of the EGFR gene, the most frequent genetic change in adult primary glioblastoma, is rarely observed in pediatric tumors.5,6 The TP53 mutations, rarely found in primary glioblastomas in adults, frequently occur in de novo pediatric tumors. Similarly, whereas pediatric glioblastomas rarely harbor mutations of the PTEN tumor suppressor gene at 10q23, they are found quite commonly in adult primary glioblastomas.7,8 However, if mutations of PTEN are observed, they are generally associated with poor prognosis in the pediatric population.9,10
• Although histologically indistinguishable, the molecular signatures of pediatric glioblastomas are considerably different from those of adult tumors.
Surgery remains the primary treatment modality of most supratentorial high-grade astrocytomas in children.11 However, the higher the grade of the tumor, the more locally infiltrative it becomes. This makes complete surgical resection practically impossible without significant damage to surrounding normal tissue. Nevertheless, radical surgical resection has been shown to improve outcome, and high-dose postoperative radiation plays a critical role in prolonging survival.7,8 Cranio-spinal radiation is typically unnecessary, as these tumors do not usually spread via cerebrospinal fluid (CSF) pathways. Although chemotherapy has not played a major treatment role historically, recent studies utilizing temozolomide, an alkylating agent with relatively good CNS penetration, have shown some promise.12
High-grade astrocytomas generally carry a very poor prognosis. Despite aggressive treatment with surgery and radiation, fewer than 50% of patients are alive 2 years after diagnosis, and long-term survival rates are low.13 Generally speaking, patients with anaplastic astrocytoma do tend to survive longer than patients with glioblastoma. After initial therapy, children should be carefully monitored with follow-up MRI exams.
Ependymomas
Predominantly an infratentorial lesion, ependymomas are well-circumscribed tumors that arise in children with an incidence peak at 6 years of age. However, approximately 30% of ependymomas arise supratentorially, most commonly from the lateral or third ventricle (60%) or from the cerebral hemisphere (40%). They account for 10% of all pediatric tumors and approximately 30% of tumors in children younger than 3 years of age. Histologically, ependymomas (WHO grade II) classically exhibit perivascular pseudorosettes of tumor cells radially arranged around blood vessels and true ependymal rosettes that form a central lumen. Anaplastic ependymomas (WHO grade III) demonstrate increased cellularity and brisk mitotic activity. Perivascular pseudorosettes are frequently encountered with microvascular proliferation and necrosis. Histopathological distinction between grades II and III is often difficult and entails frequent disagreement among neuropathologists.14 As a result, an impact of tumor histology on clinical outcome remains controversial.
Controversy
• There is debate over whether the current grading system for ependymomas is adequate for accurately distinguishing among grades or for predicting tumor behavior.
Pearl
• The extent of resection is an important prognostic indicator for children with ependymoma. Gross total resection should be attempted when possible.
Genetic aberrations are frequent in ependymomas and range from 61 to 79% of the studied populations.15 Oncogene amplifications are infrequent in ependymomas, and only single cases harbor the homozygous deletion of the CDKN2A/B gene.16 Importantly, the gain of chromosome 1q is a common finding in several studies associated with poor prognosis.15,17 In contrast, a better prognosis has been associated with the loss of 6q25.3 in patients with anaplastic ependymomas.18 One study has demonstrated frequent gains and even amplification of the EGFR locus and has confirmed that overexpression was an independent marker for poor survival in patients with grade II tumors.17 Interestingly, there is a correlation between anatomic location and type of genomic alteration, suggesting distinct genetic pathways in ependymoma. Gene expression profiling has identified specific expression signatures, demonstrating the involvement of both Notch and Hedge hog signaling pathways in intracranial ependymomas and suggests that radial glial cells are the origin for tumor formation.19
The benefit of complete surgical resection has been clearly demonstrated for pediatric ependymomas, and gross total re-section should be the first-line therapy when possible. Additional resection is justified when the postoperative imaging suggests residual disease.20 Postoperative radiation to the resection cavity has been shown to improve survival for children with ependymoma.21 In general, ependymomas are not chemoresponsive tumors. Current clinical trials are investigating the role of postradiation chemotherapy in preventing recurrence.
Children with ependymoma who have undergone gross total resection followed by focal radiation generally have a good prognosis, with approximately an 80% 5-year survival. If gross total resection is not achieved, 5-year survival dramatically worsens, with most studies reporting a drop to approximately 20 to 30%. Due to the possibility of late disease recurrence, children with ependymoma require follow-up for many years.
Choroid Plexus Tumors
Tumors of the choroid plexus are rare, comprising only about 1% to 2% of pediatric brain tumors. Although the histological classification can be ambiguous, two types of choroid plexus tumors are distinguished, the choroid plexus carcinoma (WHO grade III; Fig. 29.1) and choroid plexus papilloma (WHO grade I). Microscopically, papillomas may resemble normal choroid plexus, but may demonstrate a higher degree of columnar epithelium, nuclear pleomorphism, hyperchromasia, and sparse mitotic figures. In contrast, carcinomas are characterized by marked cytological and architectural atypia. They may be invasive, with high mitotic index and areas of necrosis. Germline mutations of the tumor suppressor gene TP53 are frequently observed, and genetic testing for Li-Fraumeni syndrome should be considered.22
The presenting signs of a choroid plexus tumor can include a bulging fontanel with accelerated head circumference, irritability, seizures, vomiting, and lethargy. Due to open fontanels in infants, these tumors can be exceptionally large at presentation. For the lower grade papillomas, surgical re-section may be the only treatment required. In the more aggressive carcinomas, gross total resection with adjuvant chemotherapy or radiation will offer the best opportunity for cure. However, the decision to deliver craniospinal radiation therapy to a population with a median age of 2 years is daunting due to long-term side effects. Postoperative chemotherapy for carcinoma does improve survival rates, and intensive chemotherapy is often used with the aim of delaying or avoiding radiation.23
Although both tumors have been shown to metastasize, the survival rate of patients with papilloma is significantly higher. However, progression from papilloma to carcinoma has been reported.24 Nevertheless, the rarity of choroid plexus tumors makes it difficult to establish exact rates of recurrence and cure. Follow-up recommendations for carcinoma survivors are similar to those for other pediatric brain tumors, with routine MRI at 3- or 4-month intervals in the first year. Neuropsychological assessments are essential for following cognitive development in this young group of patients.
Supratentorial Primitive Neuroectodermal Tumors
Supratentorial primitive neuroectodermal tumors (PNETs) (Fig. 29.2) are the most common group of malignant childhood brain tumors. They are embryonal tumors with divergent degrees of differentiation along neuronal, astrocytic, muscular, or melanocytic lines. For example, cerebral neuroblastomas (neuronal differentiation) and ganglioneuroblastomas (ganglion and neuronal differentiation) are histologically unique. Additionally, infrequent tumor types such as medulloepithelioma, ependymoblastoma, and embryonal tumor with abundant neuropil and true rosettes are delineated in the current WHO classification. These tumors are solely encountered in the cerebrum and occasionally in the suprasellar region.
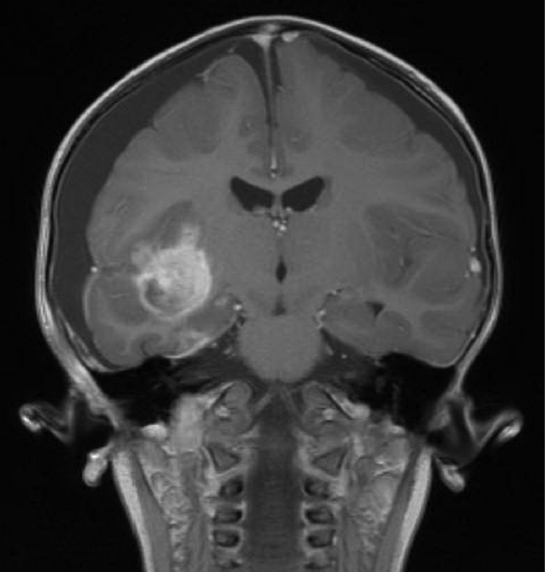
Fig. 29.2 Coronal T1-weighted image with contrast enhancement showing a right temporal primitive neuroectodermal tumor (PNET). Supratentorial PNET tends to present as a heterogeneous mass centered in deep white matter. Solid portions of the tumor tend to show isodensity to gray matter on T2-weighted images. Enhancement after contrast administration is typically heterogeneous in appearance.
• Primitive neuroectodermal tumors are typically composed of sheets of small round blue cells, and during the 1980s and 1990s there was a major controversy over whether the “small round blue cell tumors” involved distinct, location-specific entities (e.g., medulloblastomas) or whether these were different manifestations of a common underlying molecular pathway. Genome-wide profiling has since revealed specific DNA copy number alterations and messenger RNA expression signatures in supratentorial PNETs as compared with medulloblastomas. For instance, the loss of chromosome 17p, which comprises a cytogenetic hallmark of medulloblastoma, is rarely found in supratentorial PNET.
In the pediatric population, aggressive clinical behavior and the high risk of leptomeningeal dissemination are encountered in supratentorial PNETs to an extent similar to that in medulloblastomas. Thus, current treatment strategies for supratentorial PNETs are essentially based on those for medulloblastomas. However, the long-term prognosis is considerably worse than for medulloblastomas. As such, surgery plays a vital role in the treatment of all embryonal tumors, with gross total resection as the goal. The role of chemotherapy is uncertain and has not been tested in a randomized setting. Cranio-spinal irradiation followed by a boost to the primary tumor site is typically a requirement for an optimal outcome.
Atypical Teratoid/Rhabdoid Tumor
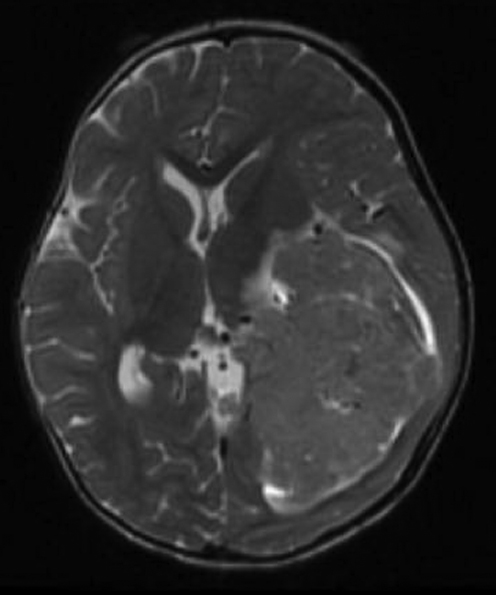
A relatively rare rhabdoid tumor occurring within the central nervous system, atypical teratoid/rhabdoid tumor (AT/RT) comprises only 1 to 2% of all pediatric brain tumors. These neoplasms typically present in children younger than 3 years of age, with approximately equal supratentorial and infratentorial distribution. Supratentorial tumors are typically located in the cerebral hemispheres (Fig. 29.3) and the sellar region. Dissemination of AT/RT via CSF is relatively common and found in 25% of patients at presentation.25 Microscopically, AT/RTs are heterogeneous lesions composed of rhabdoid cells with eccentrically placed nuclei and abundant cytoplasm with eosinophilic inclusions. Tumors may contain variable tissue components with primitive neuroectodermal, epithelial, and mesenchymal features.
Immunohistochemical staining with integrase interactor 1 (INI1) antibody shows a very sensitive and specific pattern in AT/RT. AT/RTs typically show a loss of nuclear expression, often in association with mutation or deletion of the genomic locus of the encoding gene, SMARCB1. Loss of INI1 protein is detected in almost all AT/RTs, and most tumors (75%) have detectable deletions and mutations of the INI locus. The localization of mutations within the INI1 gene appears to vary between tumors from different sites of origin, and exons 5 and 9 are considered hot spots for AT/RTs located within the CNS.26 Thus, rapid identification of these tumors is possible using immunohistochemical staining for INI1 expression supplemented with mutation analysis.27,28
Patients with AT/RT have significantly shorter survival than patients with PNET undergoing comparable therapy (mean survival less than 12 months) and often exhibit rapid progression. No difference in outcome has been observed in patients with various types of INI1 alterations, including mutations in specific INI1 exons. However, patients who are older than 3 years of age at presentation appear to have a longer survival. Current treatment protocols for AT/RT are evaluating alternative high-intensity chemotherapy and the early utilization of radiation therapy. This approach is based on the observation that long-term survivors in previous studies have often received multiagent chemotherapy, early radiation therapy, or a combination of the two.29,30
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


