Chapter Outline
PATHOLOGY OF PEDIATRIC RENAL TUMORS
Clear Cell Sarcoma of the Kidney
Congenital Mesoblastic Nephroma
Malignant Rhabdoid Tumor of the Kidney
Primitive Neuroectodermal Tumor of Kidney
Molecular Biology and Genetics
Wilms Tumor Subsets Defined by Genetic and Epigenetic Features
Clinical Presentation and Diagnostic Evaluation
CLEAR CELL SARCOMA OF THE KIDNEY
Molecular Biology and Genetics
Xp11.2 and t(6:11) (p21:q12) Translocation Carcinomas
Clinical Presentation and Diagnostic Evaluation
CONGENITAL MESOBLASTIC NEPHROMA
Pediatric renal tumors account for approximately 5% of malignancies in children younger than 15 years old and 3.6% of malignancies in children younger than 20 years old. Among 9731 patients registered with the National Wilms Tumor Study Group (NWTSG) (1969–2002), Wilms tumor (WT) composed the vast majority of childhood renal tumors (92%), followed by clear cell sarcoma of the kidney (3.4%), congenital mesoblastic nephroma (1.7%), malignant rhabdoid tumor (1.6%), and rare miscellaneous neoplasms, including primitive neuroectodermal tumor, synovial sarcoma, neuroblastoma, and cystic nephroma (1.1%). Although not historically included on NWTSG studies, renal cell carcinoma accounts for 8% of renal tumors in children from birth to 19 years, according to data from the Surveillance, Epidemiology, and End Results (SEER) program.
The study of pediatric renal tumors has had a significant impact on the field of oncology. WT has provided a paradigm for multidisciplinary treatment approaches and the conduct of cooperative group studies. WT investigators took the lead in establishing one of the leading biologic-sample banks, which now contains several thousand annotated tumor, blood, and urine samples. Tenets of cancer biology, including Knudson’s two-hit model and loss of imprinting in tumorigenesis, were pioneered in WT. This chapter reviews the pathology, epidemiology, genetics and biology, and treatment of pediatric renal tumors.
Pathology of Pediatric Renal Tumors
Staging
Stage is the major determinant of prognosis and therapy for all pediatric renal neoplasms. Two staging systems are widely used ( Table 55-1 ). The Children’s Oncology Group (COG) staging system reflects tumor extent after surgery before chemotherapy is given. By contrast, the International Society of Pediatric Oncology (SIOP) system reflects tumor extent after 4 to 6 weeks of chemotherapy and surgery. The COG and SIOP staging systems are applied to all pediatric renal tumors except renal cell carcinoma, for which the TNM (tumor, nodes, metastasis) system is used.
| Children’s Oncology Group (COG) (Prechemotherapy) | International Society of Pediatric Oncology (SIOP) (Postchemotherapy) | |
|---|---|---|
| I |
|
|
| II |
|
|
| III |
|
|
| IV | Hematogenous metastases or spread beyond abdomen | Hematogenous metastases or spread beyond abdomen |
| V |
|
|
The COG staging schema is very similar to that used in the National Wilms Tumor Study 5 (NWTS-5), although one change distinguishes the current COG staging criteria from the prior NWTS-5 criteria. Localized spillage or rupture, or any form of biopsy before removal of the kidney, is no longer considered stage II but instead is considered stage III. The SIOP system was recently revised to classify tumors with local and regional lymph node involvement as stage III. Previously lymph node involvement was considered stage II, with separate designations for node-positive and node-negative disease.
Wilms Tumor (Nephroblastoma)
Gross Features
WT typically presents as a unicentric, spherical mass that is sharply demarcated from the renal parenchyma. Approximately 10% of WTs are multicentric, a finding that is associated with an increased likelihood of WT formation in the contralateral kidney. Approximately 5% to 6% of WTs are bilateral at presentation.
The cut surface of WT is generally pale gray and uniform, but hemorrhage, necrosis, and cyst formation are common. The texture is usually soft and friable, but stroma-rich neoplasms may have a dense, myomatous consistency. Calcification is relatively uncommon. Prominent septa frequently impart a multinodular appearance.
WT may arise anywhere in the cortex or medulla, frequently compressing and distorting renal parenchyma around its margin. Rarely exophytic tumors connected to the renal surface by a narrow stalk mimic extrarenal WT. Polypoid masses in the pelvicalyceal lumen may occur either as extensions from the primary intrarenal neoplasm or as separate neoplasms arising within the pelvic wall. The renal vein and its branches not uncommonly are filled by tumor thrombus that can extend via the inferior vena cava into the right atrium.
Cystic Variants of Wilms Tumor
Scattered cysts are commonly encountered in conventional WT, but rarely the encapsulated, well-delineated neoplasm is composed entirely of cystic spaces and delicate septa, without an expansile solid component. For neoplasms that are entirely composed of mature cells, the term is cystic nephroma (CN). If the septa contain embryonal cell types, the designation cystic, partially differentiated nephroblastoma (CPDN) is appropriate. In contrast, cystic WT is distinguished by the presence of solid, expansile regions that replace or distort the cystic spaces, rather than being passively molded by the cysts. This distinction is best made by careful examination of the gross specimen.
CPDN has traditionally been thought to be a transitional stage in a continuum ranging from CN to WT. However recent studies show that CN is associated with DICER1 mutations, whereas such mutations are not observed in CPDN. This suggests that the two cystic renal tumors are distinct entities. The distinction of CN from CPDN has little clinical importance when the lesion has been completely resected because both lesions are curable by resection alone.
Microscopic Features
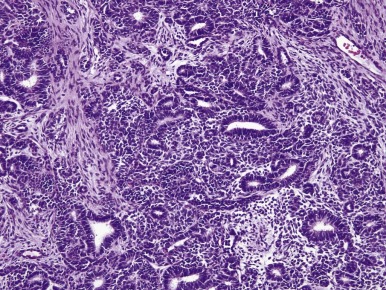
WTs are surpassed only by teratomas in their diversity of cell and tissue types and degrees of differentiation. Most WTs exhibit, at least focally, the so-called triphasic appearance, including cells of blastemal, stromal, and epithelial lineage ( Fig. 55-1 ). However monophasic and biphasic WTs are relatively common, consisting of only one or two of these cell lineages. Each of the three major cell types can demonstrate a spectrum of patterns and degrees of differentiation, accounting for the remarkable diversity of appearances characterizing various WTs.
Blastemal Patterns.
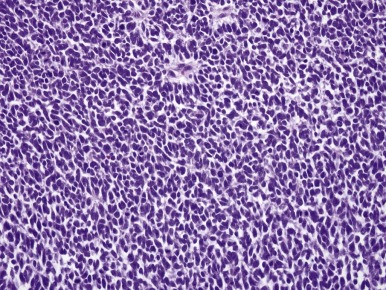
The blastemal cells of WT are small, tightly packed cells with a high nucleus-to-cytoplasm ratio, demonstrating little or no evidence of differentiation toward epithelial or stromal cell types at the light-microscopic level. Their nuclei are usually round or oval, with moderately coarse chromatin, and mold to one another in the same fashion as do the nuclei of small cell carcinoma of the lung. Nucleoli are relatively inconspicuous. Blastemal cells form several distinctive aggregation patterns, which can be divided into two broad categories, diffuse and nested, depending on their structure and degree of invasiveness. Monomorphous, relatively dyscohesive sheets of blastemal cells with aggressively invasive margins characterize the diffuse blastemal pattern. This is the most consistently aggressive pattern of WT. Stage I WTs rarely show this pattern; the majority of diffuse blastemal WT present at advanced stage, stage III or IV. Fortunately most neoplasms with the diffuse blastemal pattern are highly responsive to modern therapeutic protocols, so this pattern remains in the “favorable histology” category. The diffuse blastemal pattern can be readily confused with other small blue cell tumors of childhood, and ultrastructural or other special studies may be required to establish the correct diagnosis ( Fig. 55-2 ).
Nested blastemal patterns are characterized by sharply outlined clusters of blastemal cells in a myxoid mesenchymal background. They usually lack the invasive behavior seen with the diffuse blastemal pattern, and these neoplasms have sharply defined margins at their advancing edge. Several nested patterns may be seen. The serpentine blastemal pattern features long, serpiginous, anastomosing cords of blastemal cells in a loose, stellate, spindled stroma. This is a highly distinctive pattern of WT and helps distinguish WT from other small blue cell neoplasms of childhood. The nodular blastemal pattern resembles the serpentine pattern but has rounded blastemal nests instead of long cell cords. The basaloid blastemal pattern resembles the serpentine or nodular patterns, except that the blastemal cell clusters are outlined by a palisaded layer of cuboidal or columnar cells, reminiscent of the architecture of cutaneous basal cell carcinoma.
Epithelial Patterns.
Epithelial differentiation in WT produces a variety of cell types and degrees of differentiation. Most of these recapitulate events in normal nephrogenesis (homologous differentiation). Others, such as squamous differentiation or mucinous differentiation, do not occur in the normal kidney at any stage of development (heterologous differentiation). Tubular differentiation is the most frequent epithelial pattern. This ranges from vague hints at tubular formation in blastemal foci (which may resemble rosettes) to highly differentiated tubules resembling those of the mature kidney. WTs in which tubular differentiation is predominant tend to be less aggressive, and most of these present at stage I. Although not usually invasive, tubular-predominant WTs often grow rapidly, and it is not uncommon to find huge tumors that remain confined to the kidney. Glomerular differentiation, present in many WTs, ranges from simple papillary formations barely suggesting glomerulogenesis to mature tumor glomeruli closely resembling those of normal developing kidneys.
Stromal Patterns.
Immature myxoid and spindled mesenchymal cells are the most common stromal cell types seen in WTs. Skeletal muscle is the most common heterologous cell type. The presence of skeletal muscle (rhabdomyoblastic differentiation) in WT must not be confused with renal rhabdoid tumor, which is discussed later in this chapter. In fact, the presence of skeletal muscle in a pediatric renal neoplasm is very strong evidence supporting the diagnosis of WT.
Anaplasia: the Sign of Unfavorable Histology in Wilms Tumor
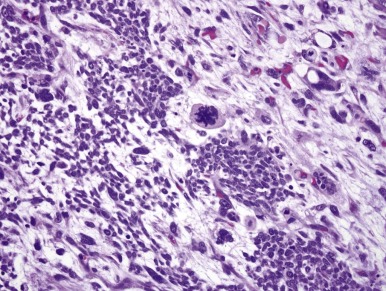
Approximately 5% of WTs demonstrate anaplastic nuclear change, the only criterion of “unfavorable histology.” All WTs lacking this feature are designated as having “favorable histology.” Anaplasia is almost never seen in WT diagnosed during the first year and is rare in the second year of life. The relative frequency of anaplasia increases after that age, and it is found in approximately 10% of WTs diagnosed after the age of 5 years. Anaplastic nuclear change reflects extreme polyploidy and is usually apparent under low magnification (10× objective). The features of anaplasia include (a) markedly enlarged tumor cell nuclei with increased chromatin content (hyperchromasia) and (b) multipolar mitotic figures ( Fig. 55-3 ). The former criteria reflect polyploidy, whereas the latter criterion helps exclude degenerative nuclear changes, as are commonly seen in cells showing skeletal muscle differentiation, from the anaplastic category. Anaplasia is tightly correlated with the presence of TP53 gene mutations. Whereas favorable-histology WT virtually never harbors TP53 mutations, the majority of anaplastic WTs do. At a practical level, p53 protein overexpression by immunohistochemistry correlates well but not perfectly with morphologically defined anaplasia.
Focal and Diffuse Anaplasia.
Initially the presence of anaplastic nuclear changes in any region of a WT was considered prognostically unfavorable. It subsequently was suggested that anaplasia is an indicator of increased resistance to adjuvant chemotherapy and not necessarily a marker of increased tumor aggressiveness; therefore stage I anaplastic WT and WT with limited intrarenal foci of anaplasia (focal anaplasia [FA]) would be predicted to have an excellent prognosis. This concept implies that the prognosis for a patient with anaplastic WT is determined by the completeness of surgical removal of anaplastic cells. However recent data on patients with stage I anaplastic WT have challenged this concept. Regardless the definition of FA includes only those WTs meeting the following criteria:
- 1.
Anaplasia is confined to one or more discrete sites within the primary tumor and is not present in extrarenal sites.
- 2.
Tumor cells outside anaplastic foci show no “nuclear unrest” (nuclear or mitotic abnormalities that approach, but do not quite attain, the degree of severity required for a designation of anaplasia).
Any WT not meeting this definition of FA is designated as having diffuse anaplasia (DA). Any of the following situations merits assignment to the DA category:
- 1.
Nonlocalized anaplastic change
- 2.
Anaplastic change or nuclear unrest in invasive sites or any extrarenal deposits
- 3.
Localized anaplastic change in a tumor that also has severe nuclear unrest elsewhere
- 4.
Anaplasia in a random biopsy specimen
- 5.
Anaplasia involving the edge of one or more sections, and the site(s) from which the sections were taken cannot be determined. (The latter emphasizes the importance of mapping the sections taken from a WT, best done on a photograph of a cut section of the tumor.)
Anaplastic foci are usually clearly demarcated from adjacent nonanaplastic tumor. Most cases in the FA category have a single focus of anaplasia. Anaplastic foci are usually only a few millimeters in diameter but can be larger if their localized nature can be convincingly documented.
The SIOP Postchemotherapy Histologic Classification System
Patients treated on SIOP studies receive several weeks of chemotherapy before undergoing nephrectomy. Chemotherapy usually affects necrosis of immature and actively proliferating cell types in WT, whereas slowly replicating and differentiated cell types are usually unaffected. For example, tumors composed mostly of mature skeletal muscle or renal tubules show little clinical regression with chemotherapy because so few cells are proliferating. The microscopic appearance of the tumor after chemotherapy has prognostic significance. Approximately 5% to 10% of WTs are completely necrotic after chemotherapy, a finding associated with a 98% 5-year relapse-free survival rate. By contrast, WTs with a predominance of blastemal cells after chemotherapy, defined as viable cells in more than one third of the tumor mass and blastemal cells in at least two thirds of the viable component, have relapse rates of nearly 40%. Based on these observations, the SIOP histologic risk classification schema divides renal tumors into three risk categories: low risk, intermediate risk, and high risk. The COG and SIOP histologic classification systems are compared in Table 55-2 .
| Children’s Oncology Group (COG) | International Society of Pediatric Oncology (SIOP) |
|---|---|
|
|
Ultrastructural and Immunohistochemical Studies
Ultrastructural study is rarely necessary to establish a diagnosis of WT but can occasionally be very helpful in distinguishing blastemal WT from other undifferentiated neoplasms. The diversity of differentiation in WT creates a correspondingly varied profile of immunohistochemical results. Blastemal cells may or may not label for vimentin and cytokeratin, whereas various differentiating elements will label according to their patterns of differentiation. For example, primitive rhabdomyoblasts within a tumor will label for the myogenic transcription factor myogenin and for cytoskeletal proteins such as actin and desmin. WT blastemal cells characteristically label for desmin but not for actin, myogenin, or other muscle markers. Immunoreactivity for WT1 protein is typically limited to the blastemal and epithelial components of WT, with the stroma being negative. Hence absence of labeling for WT1, particularly in a stroma-rich tumor, does not exclude the diagnosis of WT. Although WT1 immunoreactivity may help distinguish WT from PNET, it should be noted that desmoplastic small round cell tumor typically labels for WT1. Although useful, WT1 immunoreactivity cannot in and of itself establish or disprove a diagnosis of WT.
Nephrogenic Rests and Nephroblastomatosis
In more than 30% of kidneys resected for WT, the renal parenchyma contains one or more regions of persistent embryonal tissue, representing potential precursors of WT. These lesions have been given many names in the past; however, the term suggested by Dr. J. Bruce Beckwith, nephrogenic rests (NRs), is the currently accepted terminology. The presence of multiple or diffusely distributed NRs is called nephroblastomatosis. This term is most commonly applied when the NRs are in an active state of cellular proliferation and are large enough to be apparent on imaging studies.
NRs have a dynamic life history that can yield a variety of appearances. Dr. Beckwith’s classification is based on the assumption that the structure of NRs reflects both the dynamic state and the history of an individual rest. A brief summary is provided here.
Two fundamental categories of NRs are recognized, based on their topographic relation to the renal lobe. These are designated perilobar nephrogenic rests (PLNRs) and intralobar nephrogenic rests (ILNRs). ILNRs may occur anywhere in the renal lobe, including the peripheral cortex. They may also occur within the renal sinus, including in the walls of the pelvicalyceal system. ILNR have a more varied structure than do PLNRs and typically intercolate between nephrons, whereas PLNRs usually are discrete structures that are well delineated from adjacent nephrons. Rests of either major category may be further classified on the basis of their developmental fates. An individual NR may undergo any of the following fates, several of which often occur sequentially over time:
- 1.
It may remain unchanged in size or composition, even for many years, as a tiny, microscopic blastemal focus (dormant NR).
- 2.
It may undergo maturation, sclerosis, and eventual disappearance (sclerosing NRs and obsolescent NRs). This is the most common fate of NRs.
- 3.
It may undergo hyperplasia, or coordinated proliferation of all susceptible cells of the rest, as distinguished from a clonal neoplastic process originating in a single cell of the rest. Hyperplastic NR may produce large, actively growing masses that have numerous mitotic figures, and a section from the interior of a hyperplastic NR may be indistinguishable from WT. Several features do help make this distinction. First diffuse hyperplastic growth, involving all or most cells of a rest, tends to preserve the original shape of the rest. When PLNRs form a continuous layer of embryonal cells at the lobar surface, hyperplastic proliferation will produce a thick “rind” of abnormal tissue at the renal surface. Ovoid and lenticular masses result from hyperplasia of NRs that originally had these shapes. An irregular-shaped, multinodular appearance will result if only some of the cells are capable of proliferation. In contrast, WT, a neoplasm presumably arising from a single cell, tends to form spherical masses. The second important feature distinguishing actively hyperplastic NRs from WT is the usual absence of a pseudocapsule at the interface between hyperplastic NRs and the renal parenchyma.
- 4.
Neoplastic induction is assumed to represent a clonal event originating in single cells of a rest, resulting in WT or benign adenomas. As mentioned previously rapidly growing tumors originating at a single point (or cell) will tend to grow equally in all directions, forming spherical, expansile nodules with compressed rest remnants often present at the periphery.
- 5.
Very rarely, anaplasia may develop in nephrogenic rests.
An individual rest commonly progresses through several of these processes sequentially. For example, an incipient or dormant rest may undergo hyperplasia, followed by phases of growth arrest and maturation. This will result in a large but inactive-appearing lesion. Ultimately, one or more cells within the regressing rest may be induced to form a WT.
ILNRs are most often found at the tumor-kidney interface, where they can be misinterpreted as infiltrating tumor cells or effaced by tumor compression. A helpful feature distinguishing ILNRs at the edge of a WT is the poorly defined, irregular outer border of the ILNR, which contrasts with the sharp, pushing interface between the WT and the rest within which it arose. Also ILNRs are usually less cellular than the adjacent WT that they surround.
The presence of NRs in a kidney removed for WT is correlated with an increased risk for subsequent WT formation in the remaining kidney. The type of rest and the age of the patient modify this risk. When a carefully sampled kidney is free of rests, the risk of contralateral WT is extremely low. The possibility of subsequent WT developing in the remaining kidney should be considered in planning the follow-up of patients whose nephrectomy specimen reveals the presence of NRs in addition to WT.
Differential Diagnosis of Wilms Tumor
Triphasic Wilms Tumor.
The triphasic pattern rarely presents a problem in diagnosis, except when small biopsies are obtained from large retroperitoneal masses of uncertain origin. In this setting other mixed neoplasms might deserve consideration, including teratoma, hepatoblastoma, pancreatoblastoma, teratoma, mesothelioma, synovial sarcoma, and intraabdominal desmoplastic small round cell tumor. In the absence of nephrogenic differentiation or the distinctive nested blastemal patterns described previously, ancillary studies such as immunohistochemistry, molecular biology, or electron microscopy may be required to distinguish some of these lesions from WT. WT with extensive heterologous differentiation (“teratoid WT”) is easily confused with immature teratoma. Renal teratomas are extremely rare, and some of the reported cases likely represent teratoid WT.
Blastemal Wilms Tumor Versus Other Small Blue Cell Tumors of Childhood.
The problem of distinguishing blastemal WT from other small blue cell tumors is most likely to arise when dealing with biopsies from metastatic sites or from large abdominal tumors of uncertain origin. The distinctive aggregation patterns of blastemal WT, the presence of nuclear molding, or the focal presence of tubular differentiation will often reveal the diagnosis. Early tubular differentiation in WT lacks lumens and therefore can sometimes be confused with rosettes of neuroblastoma or primitive neuroectodermal tumor (PNET). The presence of true lumens even focally confirms tubular differentiation, whereas neurofibrils are diagnostic of a neuroblastic pseudorosette. Neuroblastic rosettes do rarely occur in WT, but usually only in teratoid WTs, which are not likely to be confused with neuroblastomas or PNETs. Immunohistochemistry, electron microscopy, molecular diagnostic techniques, cytogenetics, circulating tumor markers, and other special studies are often required to confirm the nature of a small blue cell tumor.
Epithelial Predominant Wilms Tumor Versus Papillary Renal Cell Carcinoma.
The challenge of distinguishing epithelial predominant WT from papillary renal cell carcinoma (PRCC) is most often encountered in tumors from adolescents or adults. Papillary adenoma, the benign precursor of PRCC, may resemble epithelial predominant NRs, and epithelial predominant WTs can have a predominantly papillary architecture. Sometimes PRCCs have a predominant tubular or solid component. Molecular or cytogenetic studies may be helpful because PRCC characteristically contains increased copies of chromosomes 7 and 17 and, in tumors from male patients, deletion of Y. Unequivocal glomerular differentiation, characteristic blastemal aggregation patterns, or the presence of heterologous cell types can confirm the diagnosis of WT. Immunoreactivity for cytokeratin 7 has been advocated as a useful marker for PRCC, but focal positive labeling for this marker occurs in many WTs. Only when diffusely positive is cytokeratin 7 labeling likely to be discriminatory. Nuclear labeling for WT1 protein may distinguish WT, NRs, and metanephric adenoma from PRCC, insofar as only the latter does not label.
Clear Cell Sarcoma of the Kidney
Gross Appearance
Clear cell sarcoma of the kidney (CCSK) is always unicentric, with a distinct tumor-kidney junction. Tumors are usually relatively large, with a mean specimen diameter of approximately 11 cm. The cut surface is often glistening and gelatinous. Cysts are almost always present and may be so prominent as to suggest cystic nephroma on gross examination or imaging studies.
Microscopic Appearance
Under low magnification, most CCSKs appear monomorphous, without the prominent lobulation usually seen in WT. CCSK usually has a scalloped border that appears fairly sharp under low power. Under higher magnification, the border appears less sharply defined because of the penetration of neoplastic cells a short distance into the surrounding kidney or the tumor capsule. This growth pattern tends to surround and isolate individual single nephrons, which is rarely if ever seen with WT. The entrapped tubules are usually confined to the peripheral 2 to 3 cm of a CCSK. Their epithelium commonly shows embryonal metaplastic changes similar to those entrapped in congenital mesoblastic nephroma, and the resultant basophilic epithelium creates confusion with WT. Dilation of these entrapped tubules produces intratumoral cysts that may mimic cystic nephroma. CCSK may show a wide variety of morphologic patterns, described in the subsequent sections.
Classic Pattern.
The hallmark of the classic pattern is an evenly distributed network of vascular septa, which has a branching, “chicken-wire” pattern similar to that seen in myxoid liposarcoma or oligodendroglioma. These fibrovascular septa subdivide the tumor into a conspicuous pattern of cords and nests, averaging six to ten cells in width, composed of polygonal cells that usually lack distinct cytoplasmic borders. Cells within the cords are less densely packed than those of blastemal WT, and overlapping nuclei are less frequent. Nuclear chromatin is usually finely granular, with inconspicuous nucleoli ( Fig. 55-4 ). In well-fixed specimens, the fine nuclear chromatin pattern is the most helpful clue to the diagnosis. However, it is influenced markedly by the timing and type of fixation. Mitotic figures are variable in number but are usually less numerous than those in WT. The cytoplasm usually lacks distinct borders and is surrounded by extracellular mucopolysaccharides, which creates the illusion of clear cytoplasm.
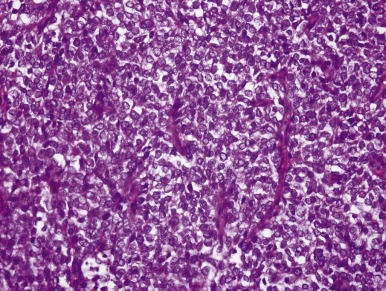
CCSK is easily confused with other neoplasms because the classic pattern is often modified, presenting alterations that mimic other neoplasms to a sometimes striking degree. Fortunately the classic pattern of CCSK predominates in most specimens and is present at least focally in more than 90% of tumors. However the pathologist who is unaware of these variant patterns is likely to diagnose CCSK as another neoplasm, with different therapeutic and prognostic implications. These variant patterns are described in the subsequent sections.
Epithelioid Patterns.
Condensation of cord cells of the classic CCSK pattern creates striking epithelioid arrangements in approximately 15% of cases. These condensations usually form trabeculae or rosettes. These epithelioid formations conform to the pattern of the original cell cords and can be either straight or undulating in configuration. The epithelioid variants of CCSK are most likely to be mistaken for WT. In all of the examples analyzed by immunohistochemistry, these epithelioid formations have been negative for epithelial markers, in contrast to the epithelial cells of WT.
Spindle Cell Patterns.
Spindle cell patterns are observed in approximately 10% of cases and result from two mechanisms. Proliferation of septal cells produces wide spindle cell septa that compress or obliterate the cell cords. Intersections of these hyperplastic septal cells often resemble the storiform patterns seen with fibrohistiocytic neoplasms. Spindled transformation of cord cells yields a spindled pattern with preservation of thin fibrovascular septa.
Myxoid and Sclerosing Patterns.
Most CCSKs contain abundant mucopolysaccharide, apparently produced by the cord cells. This material separates cord cells and creates the appearance of clear cytoplasm. In approximately 30% of cases, mucopolysaccharide occupies more volume than the neoplastic cells themselves and forms large pools or cystic spaces. While it accumulates, the tumor cells become progressively more isolated and eventually degenerate. In time the mucoid material becomes denser, eosinophilic, and hyaline in appearance. Hyaline sclerosis is found in 35% of CCSK. Complete replacement of cell cords by hyaline sclerosis preserves the original cord pattern with retention of the vascular septa, preserving the chicken-wire appearance. More diffuse zones of dense stromal sclerosis surrounding individual tumor cells may create an osteosarcoma-like appearance. Dense stromal sclerosis is relatively uncommon in untreated WT, and this finding can be a clue to the diagnosis of CCSK or rhabdoid tumor in limited biopsy material.
Palisading (Verocay-Body) Pattern.
Nuclear palisading resembling schwannoma is focally prominent in approximately 10% of CCSK specimens. Unlike schwannomas, these areas are not immunoreactive for S-100 protein.
Monstrocellular (Anaplastic) Pattern.
Approximately 3% of CCSK contain foci with enlarged, pleomorphic nuclei and bizarre mitotic figures, resembling the appearance of anaplastic WTs or pleomorphic sarcomas. This is the only pattern of CCSK that frequently overexpresses p53 protein.
Posttherapy Patterns.
Recurrences of CCSK after therapy may have a deceptively hypocellular and bland appearance, suggesting low-grade fibromatosis or myxoma. A lump appearing anywhere in a child with a history of CCSK should be viewed as a potential metastasis until proven otherwise.
Ultrastructure, Immunohistochemistry, and Cytogenetics
Ultrastructural studies yield few clues as to the putative cell of origin of CCSK. The tumor cells are characterized by a high nuclear-to-cytoplasmic ratio, with nuclear shapes that are more irregular and variable than would be expected from light microscopy. The cytoplasm is usually tenuous, with elongated, irregular processes surrounding abundant intercellular matrix. This latter feature is responsible for the vacuoles often seen with the light microscope. The cytoplasm tends to be poor in organelles. Immunohistochemistry has afforded little new insight into the histogenesis of CCSK, except to exclude various potential lines of differentiation for this enigmatic neoplasm. Vimentin is positive in nearly all specimens and BCL2 in some, but other markers are consistently negative. These include stains for epithelial markers (cytokeratins and epithelial membrane antigen [EMA]), neural markers (S-100 protein), neuroendocrine markers (chromogranin, synaptophysin), muscle markers (desmin), CD34, CD117 (c-kit), and CD99 (MIC2). A subset of CCSK harbor a t(10;17)(q22;p13) chromosome translocation, resulting in a YWHAE-FAM22 gene fusion.
Differential Diagnosis of Clear Cell Sarcoma of the Kidney
The distinction of CCSK from other renal neoplasms can be extremely difficult, even for those with extensive experience in pediatric renal neoplasm pathology. The distinction of CCSK from rhabdoid tumor of the kidney is covered in the section on the latter. The major differential diagnostic concerns are discussed in the text that follows.
Clear Cell Sarcoma of the Kidney Versus Wilms Tumor.
Some blastemal WT are richly vascular, and the vascular pattern may perfectly mimic that of CCSK. Several features help in this differential diagnosis. First, under low magnification, blastemal WTs often show distinctive nested patterns, whereas CCSKs are not typically nested. Second, blastemal WTs are more densely cellular than are CCSKs. When cells are closely packed, most nuclei are overlapping, and nuclei mold to one another, WT is more likely than CCSK. Third, heterologous tissues such as skeletal muscle are never seen in CCSK. The presence of a focus resembling CCSK in an otherwise unequivocal WT can safely be ignored. Fourth, multicentricity is common in WT, whereas multicentric and bilateral CCSKs have not been reported. Fifth, sclerotic, hyalinized stroma in an untreated tumor favors CCSK over WT. Finally, a fine nuclear chromatin pattern favors a diagnosis of CCSK over WT, although this feature is susceptible to fixation and processing artifacts.
Clear Cell Sarcoma of the Kidney versus Congenital Mesoblastic Nephroma.
Cellular congenital mesoblastic nephroma (CMN) may resemble spindled variants of CCSK, and the age ranges of these two entities overlap. Small foci resembling CCSK may occur in CMN, but CMN lacks the diversity of morphologic patterns that typifies CCSK. The absence of the t(12;15) chromosome translocation that is characteristic of cellular CMN in all CCSKs examined to date indicates that these two neoplasms are unrelated.
Congenital Mesoblastic Nephroma
Gross Appearance
CMN arises unicentrically and usually appears to have arisen deep within the medial parenchyma, near the renal sinus. The renal sinus and adjacent structures on the medial side of the kidney are major sites of extrarenal spread of CMN. Surgeons and pathologists must pay particular attention to the medial margin of the CMN resection specimen. However because of the predominance of freely mobile fat in this location and retraction artifact, the medial specimen margin is notoriously difficult to evaluate, and it is rarely possible to be certain that it is free of involvement by CMN.
The appearance of the sectioned surface of CMN is variable and depends on the subtype. Classic CMNs have a tough, whorled appearance resembling that of leiomyoma, but cellular CMNs may be soft, friable tumors. Hemorrhage, necrosis, and cyst formation are present to some degree in a majority of cellular CMNs.
Microscopic Appearance
CMN is a low-grade fibroblastic sarcoma of the infantile kidney. The classic type of CMN has a fibrous, whorled gross appearance. Morphologically, classic CMN is identical to infantile fibromatosis; it is composed of bland fibroblastic or myofibroblastic cells, arranged in fascicles that dissect into the native kidney ( Fig. 55-5 ). Mitoses are usually rare, and necrosis is absent. The leading edge of the tumor characteristically induces angiomatous vascular proliferation where it abuts perirenal fat. Entrapped renal tubules may acquire a primitive appearance (embryonal metaplasia), which may cause confusion with true neoplastic tubular differentiation that would suggest a diagnosis of biphasic stromal-epithelial WT. In contrast, the cellular variant of CMN typically has a softer, more fleshy and hemorrhagic appearance with necrosis and cystification. Microscopically, cellular CMN is identical to infantile fibrosarcoma (IFS). Compared with the classic variant, cellular CMNs more often have a “pushing” border, demonstrate sheetlike growth patterns, are less often fascicular, are more cellular, and have a higher mitotic rate ( Fig. 55-6 ). Tumor cells may be polygonal with well-demarcated pink cytoplasm and nuclei with vesicular chromatin and prominent nucleoli; this raises the differential diagnosis of rhabdoid tumor of the kidney. Alternatively the tumor cells may be thinner, primitive, and spindled, yielding a more embryonal “blue cell” appearance. Mixed CMNs have areas identical to both cellular and classic CMN in varied proportions within the same tumor.
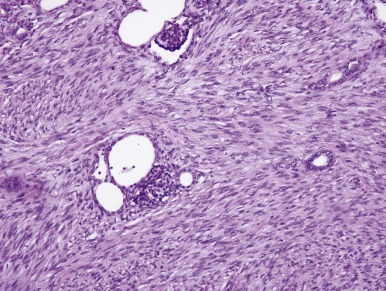
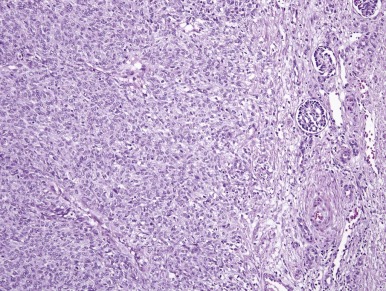
Immunohistochemistry and Ultrastructure
Immunohistochemistry is of limited value in the diagnosis of CMN. The cells of CMN label in a fashion consistent with fibroblasts or myofibroblasts. Whereas actin may be focally positive, desmin and CD34 are typically negative (P. Argani, unpublished observations). INI1 protein is retained. Ultrastructural studies also reveal features consistent with fibroblasts or myofibroblasts. Most CMN cells show abundant rough endoplasmic reticulum with branching and anastamosing profiles, and primitive cell junctions are often found.
Differential Diagnosis of Congenital Mesoblastic Nephroma
The differential diagnosis for CMN includes stromal-predominant WT, CCSK, metanephric stromal tumor (MST) and malignant rhabdoid tumor (MRT). These distinctions are crucial because most children with CMN do not require adjuvant chemotherapy, whereas those with WT, CCSK, and MRT are treated on specific chemotherapy protocols. Features that distinguish CCSK and MRT from CMN are covered in the sections on CCSK and MRT, respectively. The features that distinguish CMN from WT and MST are expanded on in the subsequent sections.
Congenital Mesoblastic Nephroma Versus Wilms Tumor.
As mentioned previously, embryonal metaplastic changes in nephrons surrounded by CMN cells are sometimes misinterpreted as tubular or papillary elements in a WT. Rarely untreated WT will be composed predominantly of stromal cells. In most of these the presence of immature or mature skeletal muscle readily excludes the diagnosis of CMN. Specimens removed after chemotherapy are particularly challenging. Treatment often ablates the embryonal, proliferating elements of a WT but tends to spare stromal cells. The resulting appearances can readily be confused with CMN. The following features are helpful in the differential diagnosis:
- 1.
A diagnosis of CMN should be considered suspect in a patient who has received prior therapy, unless the clinical and pathologic features are characteristic.
- 2.
Most CMNs are diagnosed in the first 3 months of life, whereas WT is relatively uncommon at this age. Only 10% of CMNs are diagnosed after 1 year, and virtually none are diagnosed after 2 years.
- 3.
Most WTs express a diversity of cell types and tissue patterns. In contrast blastemal foci do not occur in a CMN, and skeletal muscle is never seen. However nodules of entrapped cartilage and squamous pearls may occur in CMN (likely reflecting coexisting renal dysplasia), and this should not be taken as evidence for WT.
- 4.
The interdigitating, irregular border of classic CMN distinguished it from most WTs. However, cellular CMN often has a sharp, pushing border, similar to that of most WTs.
- 5.
Bilaterality or the presence of NRs (or both) strongly favors WT over CMN.
Congenital Mesoblastic Nephroma Versus Metanephric Stromal Tumor.
Many tumors previously considered to be CMNs in children older than age 3 years are in fact distinct from CMN and represent the newly recognized entity MST. MST is identical to the stromal component of metanephric adenofibroma (previously known as nephrogenic adenofibroma), a biphasic tumor containing an epithelial component that is identical to metanephric adenoma. MST is a benign lesion composed of spindled to stellate cells featuring thin hyperchromatic nuclei and thin indistinct cytoplasmic extensions. Several of the characteristic features of MST distinguish it from CMN. MST characteristically surrounds and entraps renal tubules and blood vessels to form concentric “onion-skin” rings or collarettes around these structures. Most MSTs induce angiodysplasia of entrapped arterioles, consisting of epithelioid transformation of medial smooth muscle and myxoid change. One fourth of MSTs feature juxtaglomerular cell hyperplasia within entrapped glomeruli, which may occasionally lead to hypertension associated with hyperreninism. Finally, unlike CMNs, MSTs are typically immunoreactive for CD34, but labeling may be patchy.
Malignant Rhabdoid Tumor of the Kidney
Gross Appearance
Most MRTs are relatively small because metastasis occurs early in the evolution of the neoplasm. On gross examination MRTs are typically fleshy or hemorrhagic neoplasms, with ill-defined borders and frequent satellite nodules that reflect the tumor’s highly invasive nature.
Microscopic Appearance
MRT was so named because of the resemblance of the tumor cells to rhabdomyoblasts. However this highly lethal infantile neoplasm does not demonstrate muscle differentiation. Microscopically these neoplasms typically engulf native renal elements at their periphery and demonstrate extensive vascular invasion. MRT classically consists of sheets of monotonous cells featuring a characteristic cytologic triad: vesicular chromatin, prominent nucleoli, and hyaline cytoplasmic inclusions ( Fig. 55-7 ). These features are variably well developed within a given tumor, which means that it may be necessary to examine multiple microscopic fields from many sections to find them.
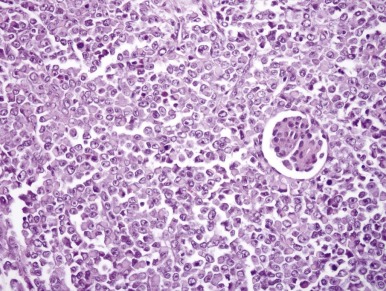
Ultrastructure and Immunohistochemistry
Ultrastructurally the cytoplasmic inclusions correspond to whorled intermediate filaments. The whorled filaments may trap antibodies used for immunohistochemistry, yielding a nonspecific polyphenotypic pattern. With this caveat, strong vimentin and focal EMA labeling are characteristic immunohistochemical findings. Recently, an immunohistochemical assay for INI1 protein has become available. Loss of INI1 protein expression reflects INI1 genetic status and, within the kidney, is a sensitive and nearly specific marker for MRT.
Differential Diagnosis of Rhabdoid Tumor of the Kidney
The diagnosis of MRT is often challenging because a wide variety of renal and extrarenal tumors of adult and children may feature fully developed “rhabdoid” cytology, at least focally. In the pediatric kidney these include cellular CMN (of the plump cell type), CCSK, blastemal WT, PNET, renal medullary carcinoma, and rhabdomyosarcoma. In general, thorough sampling of such tumors is most helpful, insofar as it typically reveals specific differentiation that excludes MRT. Additionally, the age of the patient is very helpful because MRT is not seen outside of early childhood. Finally, in the kidney loss of INI1 protein expression by immunohistochemistry is highly specific for MRT or renal medullary carcinoma. Several specific differential diagnoses are covered in the subsequent sections.
Malignant Rhabdoid Tumor Versus Wilms Tumor.
Both blastemal and myogenic elements of WT may contain cytoplasmic inclusions resembling those of MRT. A distinctive serpentine blastemal pattern, other distinctly nephrogenic features, or heterologous cells such as skeletal muscle supports the diagnosis of WT. The prominent nucleoli characteristic of MRT are not typically seen in WT, with the exception of some anaplastic specimens.
Malignant Rhabdoid Tumor Versus Cellular Congenital Mesoblastic Nephroma.
The plump cell variant of cellular CMN often has vesicular nuclei with prominent nucleoli, and a rare CMN contains foci with hyaline cytoplasmic inclusions. A predominance of spindled patterns and a less invasive tumor periphery favor a cellular CMN over a rhabdoid tumor. Cytogenetic or molecular analysis (or both) may be invaluable in this setting; demonstration of chromosome 22q loss or loss of INI1 protein expression by immunohistochemistry supports MRT, whereas the presence of the ETV6-NTRK3 gene fusion supports cellular CMN.
Malignant Rhabdoid Tumor Versus Clear Cell Sarcoma of the Kidney.
CCSK may rarely show prominent nucleoli, which can lead to difficulty in distinguishing CCSK from MRT. The presence of classic cytologic features of CCSK elsewhere and the presence of characteristic CCSK patterns are the major clues to the correct diagnosis in such instances. CCSKs lack the extreme invasiveness of MRTs and are generally more subtly invasive neoplasms.
Renal Carcinomas in Children
All of the common renal cell carcinomas (RCCs) of adulthood (clear cell, papillary, chromophobe, and collecting duct) may occasionally affect the pediatric kidney, but the clinical and differential diagnostic considerations differ in this setting. Children with von Hippel-Lindau syndrome are predisposed to develop conventional (clear cell) RCC. Children with tuberous sclerosis are at increased risk for developing both RCC and epithelioid angiomyolipoma, and immunohistochemical analysis may be needed to distinguish these two. As discussed previously, PRCC may overlap morphologically with epithelial predominant WT or metanephric adenoma. However most pediatric RCCs represent distinctive neoplastic entities. Four of these entities are described in the subsequent sections.
Xp11.2 Translocation Renal Cell Carcinomas
RCCs with chromosome translocations involving Xp11.2 and resulting gene fusions involving the TFE3 transcription factor gene were officially recognized in the 2004 World Health Organization Renal Tumor Classification. Although this subtype of RCC is relatively uncommon on a percentage basis in adults, it most likely accounts for the majority of pediatric RCCs. One distinctive subtype bears a t(X;17)(p11;q25), which results in the identical ASPL-TFE3 gene fusion, as was initially identified in alveolar soft part sarcoma (ASPS), and has been designated the ASPL-TFE3 RCC. Other subtypes are listed in Table 55-3 .
| Gene Fusion | Chromosome Translocation | Age (Years) | Tumor |
|---|---|---|---|
| ASPL-TFE3 | der(17)t(X;17)(p11.2;q25) | 5-40 | ASPS |
| ASPL-TFE3 | t(X;17)(p11.2;q25) | 2-68 | RCC |
| PRCC-TFE3 | t(X;1)(p11.2;q21) | 2-70 | RCC |
| PSF-TFE3 | t(X;1)(p11.2;p34) | 5-68 | RCC |
| NoNo-TFE3 | inv(X)(p11;q12) | 39 | RCC |
| CLTC-TFE3 | t(X;17)(p11.2;q23) | 14 | RCC |
| Alpha-TFEB | t(6;11)(p21;q12) | 6-53 | RCC |
Xp11.2 translocation RCCs closely resemble conventional (clear cell) renal carcinomas on gross examination, usually being tan-yellow and often necrotic and hemorrhagic. A papillary carcinoma composed of clear cells is the most distinctive histopathologic appearance ( Fig. 55-8 ), because this combination is uncommon in other defined types of renal carcinomas. However the Xp11.2-translocation RCCs often have nested architecture and often contain cells with granular eosinophilic cytoplasm. The histologies of Xp11-translocation RCC associated with different TFE3 gene fusions differ. The ASPL-TFE3 RCCs feature cells with voluminous, clear to eosinophilic cytoplasm, discrete cell borders, vesicular nuclear chromatin, and prominent nucleoli. In contrast, the PRCC-TFE3 RCCs, characterized by a t(X;1)(p11.2; q21), typically have less abundant cytoplasm; fewer psammoma bodies; fewer hyaline nodules; and a more nested, compact architecture.
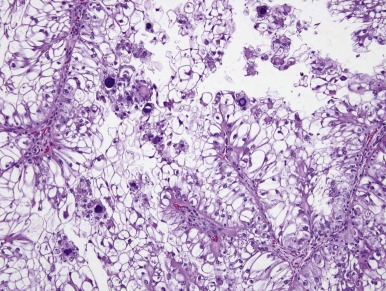
Renal carcinomas with Xp11.2-associated translocations characteristically underexpress epithelial immunohistochemical markers such as cytokeratin and epithelial membrane antigen. Only approximately one half of cases will be positive with these markers, and the labeling is often focal. Vimentin immunoreactivity is also often focal compared with that of the adjacent blood vessels; this also differs from conventional RCCs, which are diffusely positive. Rare Xp11.2-translocation carcinomas, especially ones with variant gene fusions such as PSF-TFE3 and CLTC-TFE3 , express melanocytic markers HMB45 and Melan A, creating confusion with epithelioid angiomyolipoma. Cathepsin K, a serine protease normally expressed in osteoclasts, is expressed in approximately 50% of translocation RCC but no other subtype of RCC. The most distinctive immunohistochemical feature of these neoplasms is nuclear labeling for TFE3 protein using an antibody to the C-terminal portion of TFE3, which is retained in the gene fusions. Nuclear labeling for TFE3 is a common feature of all Xp11.2-translocation RCC and ASPS but does not occur in other RCCs ( Fig. 55-9 ). Because native TFE3 is known to be ubiquitously expressed but is not detectable in normal tissues by immunohistochemistry, it is postulated that the different TFE3 gene fusions consistently lead to overexpression of the fusion protein relative to native TFE3, such that the protein becomes detectable by this assay. However TFE3 immunohistochemistry is difficult to optimize in the setting of variable fixation, and a break-apart fluorescence in situ hybridization (FISH) assay for TFE3 gene rearrangements is a more reliable assay in formalin-fixed, paraffin-embedded tissue.
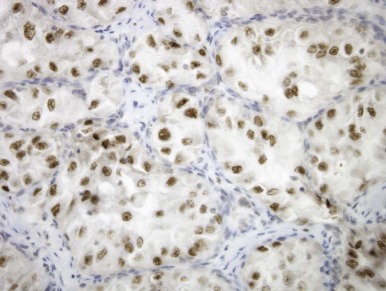
When analyzed by electron microscopy, Xp11.2-associated carcinomas feature cell junctions, microvilli, intracytoplasmic fat, and glycogen and thereby most closely resemble conventional renal carcinomas. However a subset of cases demonstrates distinctive ultrastructural features. Most of the ASPL-TFE3 RCCs contain membrane-bound cytoplasmic granules, and a few have membrane-bound rhomboidal crystals that are identical to those seen in soft tissue ASPS. Some PRCC-TFE3 RCCs have had distinctive intracisternal microtubules similar to those seen in malignant melanoma and extraskeletal myxoid chondrosarcoma.
t(6;11)(p21;q12) Renal Cell Carcinomas
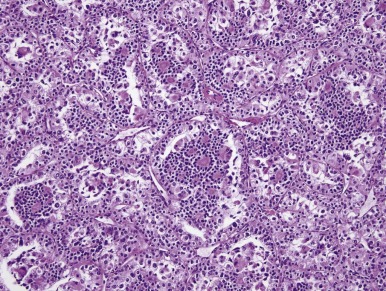
Another distinctive type of RCC bears a t(6;11)(p21;q12). The distinctive clinicopathologic features of these neoplasms were not described until 2001. On microscopic examination these neoplasms usually feature nests and tubules of polygonal epithelioid cells, separated by thin capillaries. Some cases have had papillary formations. The majority of the tumor cells have abundant clear to granular eosinophilic cytoplasm, well-defined cell borders, and round nuclei with small nucleoli. However a second population of smaller epithelioid cells is also characteristic, typically (but not always) clustered around nodules of hyaline basement membrane material within larger acini ( Fig. 55-10 ). The cases examined have generally been negative for cytokeratins by immunohistochemistry, but almost all have labeled at least focally for HMB45, Melan A, and Cathepsin K, again creating confusion with epithelioid angiomyolipoma.
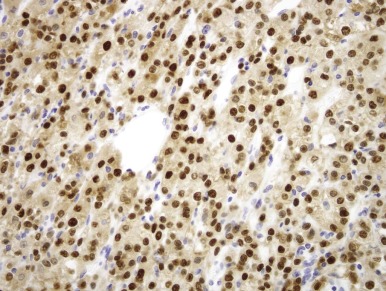
The t(6;11)(p21;q12) has been shown to result in a fusion of the intronless, untranslated Alpha gene with TFEB, a gene belonging to the same transcription factor family as TFE3 . The consequence of the Alpha-TFEB fusion is dysregulated expression of the normal full-length TFEB protein. Along these lines, the t(6;11) RCCs (also known as Alpha-TFEB RCCs) demonstrate specific nuclear labeling for TFEB protein by immunohistochemistry (IHC), whereas other neoplasms and normal tissues do not ( Fig. 55-11 ). Hence nuclear labeling for TFEB is a sensitive and specific diagnostic marker for this neoplasm with a TFEB gene fusion, just as nuclear labelling for TFE3 is a sensitive and specific marker for neoplasms bearing TFE3 gene fusions. A break-apart FISH assay for TFEB gene rearrangements has recently been developed for formalin-fixed, paraffin-embedded tissue and is proving to be more sensitive than TFEB IHC assay.
Renal Medullary Carcinoma
Renal medullary carcinoma (RMC) was first described in 1995 by Davis and colleagues as “the seventh sickle cell nephropathy.” These tumors characteristically affect young patients (mean age, 22 years) with sickle cell trait. Tumors are typically centered in the renal medulla but demonstrate an aggressively infiltrative growth pattern with frequent vascular permeation. The tumor cells feature vesicular chromatin and prominent nucleoli, frequently associated with hyaline intracytoplasmic inclusions, therefore imparting a rhabdoid cytologic appearance. The tumor cells of RMC demonstrate diffuse (sheetlike), cribriform, gland-forming, microcystic, or reticular growth patterns, the latter two simulating yolk-sac tumor ( Fig. 55-12 ). The stroma is typically desmoplastic and infiltrated by neutrophils, and close examination of extravasated red blood cells may reveal sickling. However, hemoglobin electrophorosis is more reliable than the latter for determining that the patient has sickle cell trait. These tumors frequently present at high stage and are highly lethal; Davis and colleagues reported a mean survival of only 15 weeks. Given their medullary epicenter and high-grade cytology, RMCs are considered by some to be variants of collecting duct carcinoma, although the latter entity is a notoriously elusive one to define. As noted previously, RMCs consistently show loss of expression of INI-1 protein by IHC, similar to RTK.

Oncocytic Renal Carcinomas in Neuroblastoma Patients
Meideros and coworkers have described oncocytic renal carcinomas arising in children who had previously had neuroblastoma. The genetic alterations identified in these lesions did not match those of known renal carcinomas, suggesting that these are another distinctive entity.
Primitive Neuroectodermal Tumor of Kidney
Primary renal PNET should be considered in the differential diagnosis of any undifferentiated blue cell tumor of the kidney. This neoplasm is encountered mainly in adolescent and young adult patients and tends to present at advanced stage. Tumors consist of sheets of undifferentiated small blue cells with frequent necrosis and occasional rosettes. Renal PNETs are frequently mistaken for blastemal WTs of adolescent or adult patients and might account for some of the adverse prognosis attributed to “adult WT.” Compared to WT, tumor cell nuclei are less hyperchromatic and more evenly placed. Molecular detection of the EWS-FLI1 gene fusion that results from the characteristic and specific t(11;22)(q24;q12) chromosome translocation is a useful way to confirm the diagnosis of PNET in suspected cases. Also helpful is immunohistochemical labeling for CD99; the characteristic membrane staining pattern observed in PNETs is rarely if ever observed in WT blastemal cells or cellular CCSKs, which are the main differential diagnosis.
Synovial Sarcoma of the Kidney
Similar to renal PNET, primary renal synovial sarcoma has been established as a distinctive neoplastic entity by demonstration of its specific chromosome translocation and gene fusion. Genetically confirmed cases have affected young adults. Many have previously been classified as adult blastemal WT, embryonal sarcoma of the kidney, adult cellular mesoblastic nephroma, or sarcoma arising in cystic nephroma. Renal synovial sarcomas are typically large and often cystic, and they present at advanced stage. The typical appearance is that of a monomorphic, highly cellular neoplasm composed of plump, spindled cells growing in short fascicles. Tumor cells have ovoid nuclei and minimal cytoplasm and may be associated with sclerosis. Grossly identified cysts are lined by mitotically inactive polygonal eosinophilic cells with apically oriented nuclei (“hobnailed epithelium”), and the cellularity of their walls may be deceptively low, creating confusion with cystic nephroma in limited biopsy samples. Overall, the appearances are best conceptualized as monophasic spindle-cell synovial sarcoma encircling native renal collecting ducts. True epithelial differentiation yielding a biphasic synovial sarcoma may occur, but this is less common. Immunohistochemically, tumor cells label focally for epithelial markers (EMA more likely than cytokeratin), but many cases are completely negative for all epithelial markers. Many cases have labeled for CD99. The diagnosis can be confirmed in such cases by molecular demonstration of the SYT-SSX gene fusion that results from the t(X;18)(p11;q11) that is characteristic of this neoplastic entity. As with renal PNET, renal synovial sarcoma is more aggressive than WT and also may account for some of the adverse prognosis attributed to “adult WT.”
Wilms Tumor (Nephroblastoma)
Epidemiology
The age-adjusted incidence of pediatric renal tumors in the United States is 7.5 per million children younger than 15 years of age and 6 per million children younger than 19 years of age. Although WT is the most common renal tumor overall, its relative frequency compared with other pediatric renal neoplasms varies according to patient age. Between 0 and 1 month of life, CMN is more common than WT, and after age 14 years RCC surpasses WT as the most prevalent renal cancer. In North America the average age of diagnosis for unilateral WT is 42 months for boys and 47 months for girls. Children with clinical malformations, genetic abnormalities, and tumor predisposition syndromes have a significantly earlier median age of diagnosis of WT. For bilateral WT the average age of diagnosis is 30 months for boys and 33 months for girls. The younger age of presentation for bilateral WT is due to genetic predisposition, an observation that provided the basis for Knudson’s two-hit model. The reason for the later age of presentation in girls is more elusive. An analysis from the NWTSG revealed that, compared with boys, girls had a relative excess of PLNRs, WT precursor lesions associated with genetic alterations of the Beckwith-Wiedemann syndrome locus at chromosome 11p15. Conversely boys had a relative excess of ILNRs, distinct precursor lesions associated with mutations of the WT1 gene at chromosome 11p13. ILNR-associated WTs arise at an earlier age than PLNR-associated WTs, partially explaining the gender difference in age distribution. However even in the absence of NRs, girls presented with WTs a few months later, on average, than boys. In North America there is a slight preponderance of WT in girls compared with boys, with a female-to-male ratio of 1.1 to 1.
There also are racial and ethnic differences in the predisposition to WT. Most Caucasian populations have an age-adjusted incidence of six to nine cases per million children annually. Black populations in North America and some parts of Africa exceed 10 cases per million children annually. In Japan, the Philippines, and China, the incidence is fewer than four cases per million children per year. Interestingly, in these East Asian populations, there is an excess of male subjects with WTs, the age at diagnosis is earlier than in North America, and PLNRs are rarely seen. There may also be biologic differences associated with racial differences in WT. A small study of WT in 15 Kenyan children demonstrated differences in molecular signature by imaging mass spectrometry and immunostaining compared with histology and age-matched North American controls. The Kenyan tumors exhibited more aggressive histology and worse outcome, although it is difficult to distinguish truly biologic aggressive disease from inadequate care related to resource limitations.
Several studies have uncovered maternal and paternal environmental associations with WT. Maternal exposures that have been associated with WT include cigarettes, coffee or tea, oral contraceptives, hormonal pregnancy tests, hair-coloring products, hypertension, household pesticide use, and vaginal infections. However none of these factors was confirmed by subsequent case-control studies conducted by the NWTSG. In a COG study breastfeeding was found to be associated with reduced risk of WT, although the effect was restricted to mothers with less than a college education. On the paternal side the occupations of machinist, welder, and other jobs involving exposure to hydrocarbons or lead were associated with increased risk of WT. A subsequent case-control study found an association with the paternal occupations of vehicle mechanics, auto-body repairmen, and welders, although no unifying exposure could be established. A correlation of a low Apgar score (0-5) with a higher risk (46%) of cancer diagnosis was found in a nationwide population-based study in Denmark and Sweden that followed 5,091,188 children from age 0 to 14 years from 1978 to 2006 (excluding children with birth defects). The potential impact of the low Apgar score and cancer diagnosis was largely confined to children diagnosed with cancer before 6 months of age, and the highest risk was observed for the occurrence of WT.
Molecular Biology and Genetics
Retinoblastoma and WT were the original tumors used by Knudson to establish the two-hit hypothesis of oncogenic transformation. Unlike retinoblastoma, in which alterations of a single gene ( Rb ) account for the vast majority of tumors, several genes have been implicated in the genesis of WT, and a sizable proportion of WTs have indeterminate genetic etiology. The genes that are most commonly mutated or epigenetically altered in sporadic WTs are WT1, IGF2, β-catenin ( CTNNB1 ), WTX, and TP53 . These five genes are discussed in detail in the subsequent sections, whereas other genes and loci implicated in WT are summarized in Table 55-4 .
| Locus | Gene | Gene Function | Comments | Key References |
|---|---|---|---|---|
| 11p13 | WT1 | Transcription factor | WAGR, Denys-Drash, and Frasier syndromes associated with WT predisposition; somatic mutations in 10% to 20% of WT | |
| 11p15 | WT2 (IGF2/ H19) | Insulin-like growth factor pathway | Beckwith-Wiedemann syndrome associated with WT predisposition; somatic genetic or epigenetic alterations seen in 70% of WT | |
| Xq11 | WTX (AMER1/FAM123B) | Wnt signaling pathway | Osteopathia striata congenita with cranial sclerosis syndrome has not been associated with WT predisposition but somatic mutations seen in 20% to 30% of WT | |
| 3p21 | CTNNB1 (β-catenin) | Wnt signaling pathway | Somatic mutations seen in 15% of WT; strong correlation with WT1 mutations | |
| 17q12-q21 | FWT1 | Unknown | Locus associated with familial WT; gene not identified | |
| 19q13 | FWT2 | Unknown | Locus associated with familial WT; gene not identified | |
| 17p13 | TP53 | Cell cycle checkpoint and apoptosis | Li-Fraumeni syndrome associated with WT predisposition; somatic mutations seen in 75% of anaplastic WT but uncommon in favorable-histology WT | |
| 13q12 | BRCA2 (FANCD1) | DNA repair | Fanconi anemia; biallelic mutations associated with WT predisposition | |
| 16p12 | PALB2 (FANCN) | DNA repair | Fanconi anemia; associated with WT predisposition | |
| 15q26 | BLM | DNA helicase | Bloom syndrome; associated with WT predisposition | |
| 15q15 | BUB1B | Mitotic spindle checkpoint | Mosaic variegated aneuploidy; associated with WT predisposition | |
| Xq26 | GPC3 | Wnt signaling pathway | Simpson-Golabi-Behmel syndrome; associated with WT predisposition | |
| 2q37 | DIS3L2 | Micro-RNA processsing | Perlman syndrome; associated with WT predisposition | |
| 7p14-15 | POU6F2 | Transcription factor | None | |
| 1q25-q32 | HRPT 2 (parafibromin) | RNA processing; histone modification | Hyperparathyroidism-jaw tumor | |
| 6q21 | HACE1 | Ubiquitin ligase | None | |
| 2p24 | MYCN | Transcription factor | Somatic gene amplification seen in 5% to 10% of WT | |
| 4q31 | FBXW7 | Ubiquitin ligase | Somatic mutations seen in 4% of WT; germline mutation described |
WT1
WT1 was the first gene found to be specifically mutated in WT. The gene was originally identified via study of patients with the WAGR syndrome. Children with WAGR suffer from the constellation of W T, a niridia, g enitourinary abnormalities, and mental r etardation. Chromosomal analysis of these patients revealed heterozygous constitutional deletions at 11p13. Subsequent investigation of the genes contained within these deletions identified disruption of the PAX6 gene as the basis of aniridia and WT1 as the key tumor suppressor. The remaining allele of WT1 is mutated in the WTs from these patients.
WT1 encodes an approximately 45-kd DNA-binding protein that contains domains rich in glutamine and proline in the amino terminus and four zinc fingers in the carboxy terminus, collectively suggesting that it acts as a transcription factor. The WT1 gene is quite complex insofar as it encodes numerous distinct isoforms and contains an evolutionarily conserved cryptic splice site that can result in the insertion of three amino acids, lysine, threonine, and serine (KTS), between zinc fingers 3 and 4. Notably distinct functions have been identified for the different KTS isoforms. Patients with Frasier syndrome, characterized by male pseudohermaphroditism, progressive glomerulopathy, and an increased risk of WT, have a congenital donor splice-site mutation in WT1 that results in loss of the +KTS isoform. Furthermore mice that express only the +KTS or only the −KTS isoform have distinct phenotypes.
Congenital point mutations in WT1 are also the basis of Denys-Drash syndrome. Patients with this syndrome suffer from renal failure, pseudohermaphroditism, and a susceptibility to WT. Denys-Drash syndrome is caused by missense mutations in WT1 that disrupt zinc finger binding to DNA. The phenotype of patients with Denys-Drash is quite severe, and evidence suggests that these mutations may lead to a dominant-negative protein that impairs function of the normal protein encoded by the remaining allele. Nonetheless the remaining WT1 allele is mutated in the WTs that arise in these patients, suggesting that mutation of both alleles is required for tumor formation. Notably whereas mutations in WT1 are the basis of several syndromes that predispose to the formation of WT, WT1 is mutated in only 10% to 20% of sporadic tumors. Mouse models have provided insight into the contributions of WT1 to development and tumor suppression. WT1 is expressed at high levels in the developing kidney, gonads, spleen, and mesothelium and at lower levels in several other tissues. Mice deficient for WT1 die midgestation with abnormalities of the metanephric blastema, gonads, mesothelium, heart, and lungs. Cells from the metanephric blastema in these mice undergo apoptosis, and consequently the metanephric kidney never develops. Based on these data it has been postulated that the WT1 protein regulates gene expression pathways required for morphogenesis of the kidney, and that its disruption results in aberrant stimulation of tissue-specific growth. This role in differentiation may also explain why WT is largely a disease of early childhood insofar as kidney morphogenesis is initiated in utero and is completed in early postnatal life. Indeed the level of WT1 plummets within a few weeks after birth in mice. Whereas WT1 −/− mice are not viable, WT1 +/− mice live but do not develop WTs. This differs from the human condition in which individuals with germline WT1 deletions or mutations have a greatly increased risk for developing WT compared with the general population.
β-catenin (CTNNB1)
The Wnt signaling pathway regulates the cellular processes of proliferation, differentiation, motility, and survival. Central to the pathway is the transcriptional activator β-catenin, which is actively degraded in the absence of Wnt signaling. Activating mutations in the β-catenin (CTNNB1) gene occur in approximately 15% of WTs. Interestingly mutations in β-catenin occur predominantly in tumors in which WT1 is also mutated, suggesting that these two events cooperate in the genesis of WT. In the developing kidney Wnt4 is an inducer of the mesenchymal to epithelial transition that underlies normal nephron development; disruption of normal renal development may be the means by which β-catenin mutation contributes to the formation of WT.
WTX (AMER1/FAM123B)
WTX was discovered by an array comparative genomic hybridization screen that identified deletions of the Xq11.1 locus in male patients with WT. Further investigation found deletions and mutations in girls. Intriguingly WTX is inactivated by a single hit affecting either the sole WTX copy in male subjects or the copy located on the active X chromosome in female subjects. According to the initial report, WTX was deleted or mutated in approximately one third of WTs, although other groups have reported a lower mutation frequency. Germline WTX mutations have been observed in individuals with osteopathia striata congenita with cranial sclerosis, a rare slerosing bone dyspasia. Individuals with osteopathia striata congenita with cranial sclerosis do not have increased risk for WT or other cancers, but NRs have been observed in one individual with a germline WTX mutation.
WTX is alternatively spliced to encode two isoforms of a protein with disparate functions. The short isoform of the protein is nuclear and forms a complex with β-catenin, AXIN1, APC, and β-TrCP2, ultimately promoting ubiquitination and degradation of β-catenin, thereby acting as a negative regulator of the WNT signaling pathway. The full-length WTX protein binds to the adenomatous polyposis coli protein and regulates its distribution between microtubules and the plasma membrane. WTX also interacts with WT1 protein and enhances its function as a transcriptional activator. It was recently shown to bind p53 and modulate p53 function through its activator CBP/p300.
Although WT1 and β-catenin mutations are highly concordant, there is no clear association between WT1 and WTX mutations. This suggests that WTX and β-catenin mutations are not functionally equivalent even though both proteins reside within the WNT pathway.
IGF2
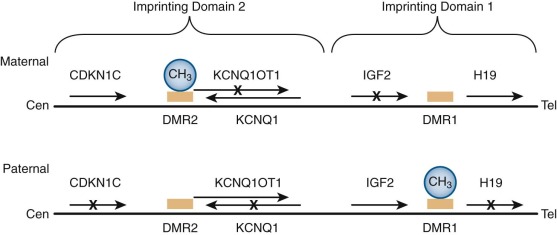
Cytogenetic analyses have long suggested the presence of an additional WT gene at 11p15 distinct from the WT1 locus at 11p13. Loss of heterozygosity (LOH) at 11p15 occurs in some cases of sporadic WT. Additionally, this locus is linked to Beckwith-Wiedemann syndrome (BWS), an overgrowth disorder associated with macroglossia, umbilical hernia, gigantism, neonatal hypoglycemia, and predisposition to WT and other cancers. BWS arises from alterations in several different genomically imprinted genes at the 11p15 locus. Genomic imprinting is the preferential expression of a gene from either the maternal or paternal allele. The molecular basis of genomic imprinting is epigenetic modification, most notably DNA methylation. The 11p15 locus has two imprinting domains ( Fig. 55-13 ). The telomeric domain (domain 1) includes the genes insulin-like growth factor 2 (IGF2) and H19 . IGF2 encodes a growth factor expressed at high levels in the developing kidney and WT. H19 encodes a biologically active untranslated RNA that may function as a tumor suppressor. In normal cells these genes are oppositely expressed such that IGF2 is expressed only from the paternal allele and H19 is expressed only from the maternal allele. The centromeric imprinting domain (domain 2) contains several imprinted genes, including KCNQ1 ( KvLQT1 ) , KCNQ1OT1 (LIT1 ) , and CDKN1C ( p57 KIP2 ). KCNQ1 encodes a subunit of a voltage-gated potassium channel that is implicated in several cardiac arrhythmia syndromes, including long-QT syndrome 1. KCNQ1OT1 encodes a noncoding RNA with antisense transcription of KCNQ1 . CDKN1C encodes a member of the cyclin-dependent kinase inhibitor family of proteins that negatively regulates cell proliferation and is a tumor suppressor. Molecular subgroups of BWS include those with (1) loss of methylation in domain 2, which is associated with decreased expression of CDKN1C (∼50%); (2) duplication of domains 1 and 2 of the paternal allele with no maternal contribution (uniparental isodisomy) (∼20%); (3) mutation of CDKN1C (∼10%); (4) gain of methylation of domain 1, resulting in increased IGF2 expression and decreased H19 expression (∼2% to 7%); 5) small duplications or chromosome rearrangements (<2%); and (5) unknown genetic etiology (∼13% to 15%). Interestingly, the Silver-Russell syndrome, associated with growth retardation, is caused by loss of methylation at domain 1 and maternal 11p15 duplication (i.e., the opposite of BWS). ,
In the past decade correlations between BWS genotypes and cancer predisposition have emerged. Uniparental isodisomy and gain of methylation at domain 1 carry the highest risk of WT. Loss of methylation at domain 2 is associated with omphalocele, macrosomia, and a distinct lack of WT, although these individuals develop other tumor types. A common thread in the patients most susceptible to WT is increased expression of IGF2 .
Wilms Tumor Subsets Defined by Genetic and Epigenetic Features
A traditional model of WT development purports that WTs fall into two discrete subtypes: type I and type II. Type I WT is caused by WT1 mutation, resulting in intralobar NRs, which then accumulate second hits such as Wnt activating mutations (β-catenin or WTX mutations). Type II WT is caused by genetic or epigenetic changes in the 11p15 locus that result in IGF2 overexpression, perilobar NRs, and presumably other second-hit genetic changes that lead to WT. Comprehensive mutation analyses of WT1, β-catenin, WTX, and the 11p15 locus in WT samples indicate that whereas some tumors fit the classic paradigm, other tumors have evidence of both IGF2 overexpression and WT1 mutation. Indeed transgenic mice with Igf2 overexpression and Wt1 deletion developed WT, whereas neither genetic abnormality alone was sufficient to cause WT. Other WTs lack evidence of either IGF2 or WT1 alteration. The requirement for coexisting mutations likely depends on the developmental stage of the cell of origin of a particular tumor.
TP53
Constitutional mutations of the TP53 gene are responsible for Li-Fraumeni syndrome (LFS), which predisposes carriers to cancer. TP53 , one of the most commonly mutated genes in human cancer, encodes a transcription factor that is a master regulator of cell cycle arrest, apopotosis, and DNA repair in response to a variety of stimuli. Although WT is not a defining criterion for LFS, several LFS families with WT have been identified. It is important to note that TP53 mutations are associated with the anaplastic histology subtype of WT; approximately 75% of anaplastic WTs have detectable TP53 mutations, whereas TP53 mutations are uncommon in favorable histology tumors. Several lines of evidence suggest that anaplastic WT arises from the acquisition of a TP53 mutation in a favorable histology WT. First, patients with bilateral WT often have discordant histologies between contralateral tumors. Second, some WTs have favorable histologic features at diagnosis but are found to have anaplastic histology at relapse. Finally, microdissected WTs were found to have TP53 mutations restricted to areas of anaplasia within the tumor tissue.
Clinical Presentation and Diagnostic Evaluation
The most common presenting signs and symptoms of WT are abdominal mass (75%), abdominal pain (28%), hypertension (26%), gross hematuria (18%), microscopic hematuria (24%), and fever (22%). The hypertension, which can be severe, may be due to ectopic renin production by the tumor, compression of the renal artery by the tumor with resulting physiologic renin production, or ectopic adrenocorticotropic hormone production. An analysis of 31 patients with WT with hypertension at diagnosis found increased plasma renin levels in 81% of patients. Some patients have persistent hypertension after the tumor has been removed, suggesting underlying renal pathology. There are no tumor markers pathognomic for WT, although associated laboratory findings include hematuria and anemia. An association between WT and acquired von Willebrand disease has been described.
WT can spread locally or hematogenously. Locally the tumor has the potential to spread into structures surrounding the kidney and can occasionally invade other organs such as the liver. If the tumor ruptures, peritoneal tumor deposits can be detected. WT also spreads to lymph nodes near the renal hilum and in the paraaortic chain. Between 6% and 8% of WTs spread through the renal vein and form a tumor thrombus in the inferior vena cava, which can propagate into the heart. The most common sites of distant metastasis are the lung and liver. There are reports of WT spread to brain or bone, but this is very uncommon. About 7% of patients have synchronous or metachronous bilateral tumors, and 10% have unilateral multifocal tumors.
Radiologic evaluation of WT focuses on the identification of local and distant sites of spread. A computed tomography (CT) scan of the abdomen and pelvis is recommended to visualize the primary tumor, the contralateral kidney, lymph nodes, and intraabdominal or pelvic tumor deposits. CT also provides information valuable to surgical planning. Assessment of the diagnostic performance of CT in identifying the presence or absence of preoperative rupture found moderate specificity but relatively low sensitivity when CT findings were compared to operative notes or pathologic examination. The single best indicator of rupture was the finding of ascites beyond the cul-de-sac, followed by perinephric fat stranding and retroperitoneal fluid. Doppler ultrasound has historically been used as a complementary test to evaluate for tumor thrombus in the renal vein and inferior vena cava. The detection of tumor thrombus by CT versus ultrasound was compared in a study population drawn from the first 1015 patients enrolled on the COG AREN03B2 Renal Tumor Biology and Risk Classification study. CT scans from 62 children with and 111 children without tumor thrombus identified at nephrectomy were reviewed. Comparable sensitivity and specificity of CT and ultrasound were found for detection of cavoatrial thrombus. These findings suggest that CT can adequately identify tumor thrombus that will affect surgical approach and that Doppler ultrasound evaluation is not a diagnostic requirement. CT imaging can also aid in surgical planning by allowing an estimation of tumor weight before surgery. In the COG AREN03B2 risk-stratification process, tumors less than 550 gm are considered for treatment with observation only, and preoperative weight estimation can be used in determining the appropriateness of central line placement at time of nephrectomy. On the other end of the spectrum, tumors greater than 1000 gm have also been associated with an increased risk of intraoperative spill; therefore estimation of larger size can also be a factor in surgical planning.
Chest x-rays have historically been used to assess patients for lung metastases, but CT scans are more sensitive in detecting nodules or effusions. On NWTS-5, 129 patients had lung lesions detected on CT scan but not chest x-ray. Forty-two of these patients with “CT-only” nodules had lung biopsies, and tumor was confirmed in 31 patients (73.8%). Retrospective studies of the prognostic significance of CT-only lung nodules have yielded conflicting results. The largest analysis to date included 186 patients with CT-only nodules from NWTS-4 and -5. These patients were treated heterogeneously (+/− doxorubicin, +/− lung radiation) because the treatment decisions were left to the discretion of the local physicians. Outcome analysis found that patients with CT-only nodules who were treated with doxorubicin had superior relapse-free survival compared with patients who did not receive doxorubicin, but the study found no impact on overall survival. The use of lung radiation did not have a significant effect on outcome. Based on these findings, the first generation of COG renal tumor studies recommended CT scans, but not chest x-rays, for the diagnostic staging evaluation for WT. Lung nodules are considered to contain tumor unless biopsy proves otherwise.
Bone scan and brain magnetic resonance imaging are not routinely recommended in the evaluation of patients with newly diagnosed WT. Fluorodeoxyglucose (FDG) positron emission tomography (PET) scanning is not a routine part of the WT workup. Initial experience with PET scans indicates that most WTs are FDG avid. Correlations of PET avidity and measures of standardized uptake value with histology and treatment response have been reported in small patient series. These studies suggest that FDG PET may have value in assessing response to chemotherapy as well as a role in identifying tumor viability in lesions with no radiologic evidence of change.
Treatment
The treatment approach to WT has evolved along two distinct pathways. The NWTSG, which became the Renal Tumor Committee of COG in 2002, advocates the upfront nephrectomy approach whenever surgically and medically feasible, followed by chemotherapy. This approach provides for accurate histologic diagnosis and staging information. In contrast, SIOP uses preoperative chemotherapy, which shrinks the tumor, thereby reducing the risk of surgical complications such as intraoperative tumor spillage. It is difficult to compare the two approaches side by side because of differences in the staging systems and treatment plans, but both approaches produce excellent outcomes. Regardless of the timing of initial surgery, the three pillars of WT treatment are surgery, chemotherapy, and radiation therapy.
Surgery
In the SIOP treatment protocols, patients 6 months of age or older receive chemotherapy before surgery unless there is evidence of tumor rupture, in which case they undergo immediate nephrectomy. Open or needle biopsy is not routinely performed. Infants younger than 6 months old are treated with upfront nephrectomy because non-Wilms renal tumors, such as mesoblastic nephroma or malignant rhabdoid tumor, are prevalent in this age group.
In the COG treatment protocols, all patients are considered candidates for immediate surgery, although there are several indications to give preoperative chemotherapy: (1) bilateral WT; (2) tumor thrombus in the inferior vena cava above the level of the hepatic veins; (3) tumors that invade adjacent organs such that resection of the tumor would involve resection of the other structures (except the adrenal gland); (4) tumors that, in the surgeon’s judgment, would result in significant morbidity or mortality if resected before chemotherapy; and (5) respiratory distress resulting from extensive pulmonary metastatic disease. If preoperative chemotherapy is given, it is recommended not to delay definitive surgery beyond week 12 of treatment.
A generous transabdominal, transperitoneal, or thoracoabdominal incision is recommended for adequate exposure. An increased rate of rupture and complication is associated with a midline laparotomy. Complete exploration of the abdomen should be performed, followed by a radical nephrectomy, dividing the ureter as distally as possible. Routine exploration of the contralateral kidney is not recommended if imaging is satisfactory and does not suggest a bilateral process because the chance of detecting a contralateral kidney tumor during surgery that is undetected with modern imaging techniques is less than 1%, and the outcome of these patients is excellent. An important component of WT surgery is lymph node sampling, even if the nodes appear normal. NWTS-4 showed that patients with positive lymph nodes had better outcomes than patients whose lymph nodes were not sampled, indicating that some patients who did not undergo lymph node sampling were understaged and undertreated. Another important facet of surgery is to remove the tumor en bloc, avoiding tumor spillage. Intraoperative spillage was shown to increase the risk of tumor recurrence. In a review of the first 2000 patients enrolled on COG Biology and Risk Stratification study AREN03B2, an intraoperative rupture rate of 9.7% was found in 1131 primary nephrectomies for unilateral WT. Risk factors for intraoperative spill were diameter (>12 cm) and laterality (right).
Surgical resection or biopsy of lung nodules plays an important role in the diagnosis of metastatic WT. Current COG renal tumor studies consider circular lung nodules, regardless of size, to be metastatic disease unless proven benign by biopsy. The resection of lung metastases may also provide a therapeutic benefit. In the SIOP protocols, patients whose lung nodules are resected completely do not receive pulmonary radiation therapy (XRT). The COG AREN0533 study tested a new strategy for favorable-histology WT with lung metastases. Lung nodules were assessed after 6 weeks of chemotherapy and XRT omitted for patients with a complete response. With this approach it is appropriate not to resect all lung nodules upfront to assess chemosensitivity.
Radiation Therapy
Radiation therapy is a mainstay of WT treatment. Initially used in all patients with WT, successive studies from the NWTSG and SIOP demonstrated that only patients with stage III or IV disease require radiation therapy ( Table 55-5 ). Among these patients radiation doses have been reduced over time, thereby decreasing the potential for late adverse effects.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


