28 Pediatric Posterior Fossa Tumors
More than half of pediatric brain tumors are located in the posterior fossa. These neoplasms encompass some of the most favorable, and some of the worst, primary brain tumors seen in children. Complete surgical removal of a cerebellar astrocytoma virtually guarantees a cure. In contrast, a medulloblastoma patient can go on to expire after gross total resection, craniospinal irradiation, and chemotherapy. The four most common lesions seen in the posterior fossa of children are medulloblastoma, ependymoma, cerebellar astrocytoma, and less commonly brainstem glioma; the first three are discussed in this chapter, and the fourth was discussed in Chapter 27.
 Medulloblastoma
Medulloblastoma
Medulloblastoma is the most common malignant brain tumor of childhood and comprises up to 18% of all pediatric brain tumors.1 Although it used to be regarded as a single heterogeneous entity, current consensus recognizes four biologically distinct subgroups—WNT, SHH, Group 3, and Group 4—with distinct genomic, clinical, and prognostic features (Table 28.1).2–4 Across subgroups, the mean age of medulloblastoma patients is between 5 and 7 years; more than half of medulloblastomas occur in the first 10 years of life but are uncommon under the age of 1 year. There is a male preponderance in most reported series (M/F ratio 1.8:1). Most cases have no known cause, but rare examples do arise in the setting of familial syndromes such as Turcot’s syndrome (polyposis of the colon and primary brain tumors), Gorlin’s syndrome (multiple cutaneous basal cell carcinomas, congenital anomalies, and medulloblastoma), and Li-Fraumeni syndrome.5 Isochromosome 17q is seen commonly in Group 3 and Group 4 and MYC amplifications in Group 3 medulloblastomas.2,6 Thus, these somatic copy number alterations are useful to identify subsets of patients with poor prognosis.7–10 In contrast, WNT tumors are associated with a good prognosis and frequently display monosomy 6, and CTNNB1 mutations/nuclear positivity.2,11
Clinical Presentation
Most patients present with a short history; in the Toronto Hospital for Sick Children (HSC) experience, symptoms had been present for less than 1.5 months in 51% of patients and less than 3 months in 76%.12 Early symptoms of a posterior fossa medulloblastoma include behavioral changes such as lethargy, irritability, and loss of appetite. These nonspecific symptoms are often not initially diagnosed as secondary to a posterior fossa tumor, and children often have extensive investigations before the correct diagnosis is finally made. Most patients present with symptoms due to increased intracranial pressure or compression of surrounding neural structures. The most common presentation is the “midline triad” of headache, lethargy, and vomiting.13 Headache is often present on awakening in the morning due to hypoventilation during sleep and a consequent rise in carbon dioxide levels, causing increased intracranial pressure. Vomiting often relieves the headache due to accompanying hyperventilation and reduction in intracranial pressure.
Pearl
• Fewer than 5% of medulloblastoma patients present due to symptomatic metastases. Presentation due to involvement of the cauda equina by “drop mets” is very unusual, but children who present de novo with multiple intradural lesions compressing the spinal cord or cauda equina should have posterior fossa imaging to rule out a cerebellar neoplasm.
Cerebellar signs such as truncal ataxia (62%), limb ataxia/dysmetria (44%), and nystagmus may be present. Many children have papilledema at the time of diagnosis. Sixth nerve palsies may be seen and are usually due to hydrocephalus and increased intracranial pressure rather than direct brainstem invasion. Facial or bulbar palsies, when present, do suggest brainstem invasion. Head tilt secondary to impaction of the cerebellar tonsils into the foramen magnum with compression of the C1 and C2 nerve roots is commonly seen in patients with cerebellar neoplasms. Infants with open sutures may have atypical presentations and can present with asymptomatic head enlargement.
There is no pathognomonic symptom or sign in patients with medulloblastoma that differentiates them from children with other types of cerebellar neoplasms. However, neck stiffness, intense neck pain, and vomiting in the absence of headache are more typical of ependymoma; lateral cerebellar signs are more common in patients with cerebellar astrocytoma. Clinically evident spontaneous tumor hemorrhage can be seen in both primary and recurrent tumors (5.6% of patients in the HSC series).12

Medulloblastoma can spread along cerebrospinal fluid (CSF) pathways and also rarely to systemic sites (bone, lung). Any child with a known medulloblastoma who presents with bone pain must have a metastatic lesion ruled out.
Imaging Studies
Classic features of a medulloblastoma on computed tomo-graphic (CT) scan are increased density on the noncontrast scan, midline location, well-defined margins, and dense, homogeneous enhancement with injection of contrast14 (Fig. 28.1). The hyperdensity on plain CT scan, seen in medulloblastoma and in some ependymomas, is secondary to the high cellularity of these tumors that have scanty cytoplasm and areas of desmoplasia.5 The tumor is usually in the cerebellar vermis (85%) but can be found in the cerebellar hemisphere (more commonly the SHH subgroup), or, rarely, in the cerebellopontine angle. Calcification is seen in 7 to 10% of cases, and true macrocysts are uncommon. Almost all medulloblastomas enhance in children, but, on occasion, adult medulloblastomas will not take up contrast. Heterogeneity of enhancement within the tumor is due to regions of necrosis.
Special Consideration
• Any patient with a posterior fossa tumor that might be medulloblastoma or ependymoma should undergo preoperative imaging of the spinal axis to rule out leptomeningeal spread. In addition to prognostication, the absence or presence of metastatic disease may prompt the surgeon to be more, or less, aggressive, respectively, at the time of resection. Patients who do not undergo preoperative spinal imaging need to wait at least 2 weeks postoperatively to facilitate optimum interpretation of imaging.
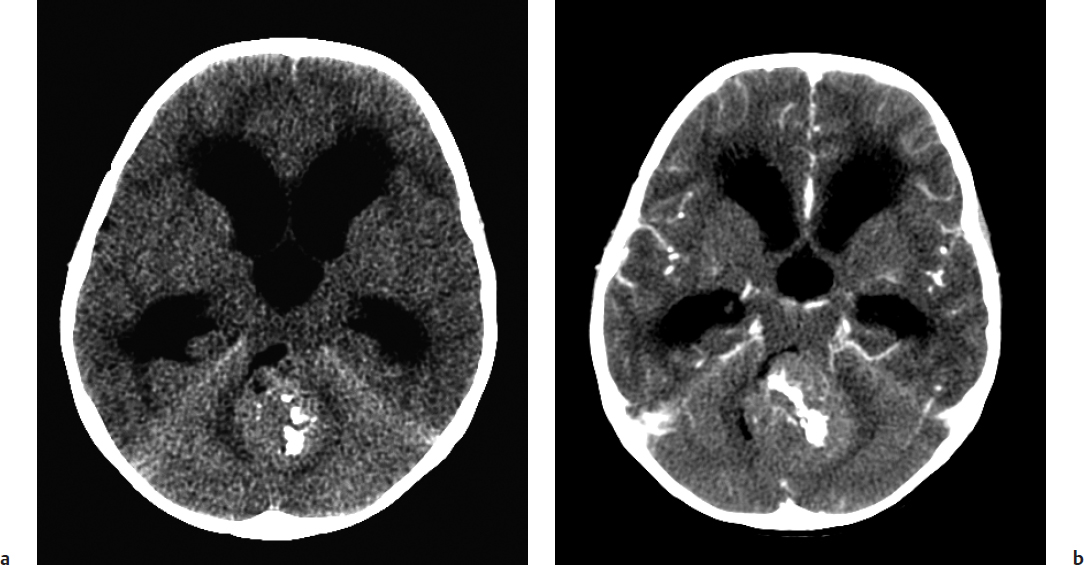
Fig. 28.1a,b (a) Non–contrast-enhanced computed tomography (CT) of a medulloblastoma showing a hyperdense mass occupying and obstructing the fourth ventricle. Note the extreme enlargement of the lateral ventricles consistent with noncommunicating hydro-cephalus. (b) Contrast-enhanced CT showing diffuse enhancement of the lesion with administration of contrast.
Obstructive hydrocephalus with enlargement of the lateral, third, and rostral fourth ventricles is extremely common on preoperative imaging. Tumor signal on T1-weighted magnetic resonance imaging (MRI) shows low or intermediate signal compared with adjacent white matter. T2-weighted MRI is varied and can be hypo-, iso-, or hyperintense to surrounding white matter (Fig. 28.2). Medulloblastomas display heterogeneous enhancement with infusion of gadolinium with lower apparent diffusion coefficient values compared with ependymomas due to relatively higher cellularity in medulloblastomas. MRI demonstrates the extent of the tumor; medulloblastoma is less likely than ependymoma to extend through the exit foramina of the fourth ventricle into the subarachnoid space of the upper cervical spinal cord or into the cerebellopontine angle. Intratumoral hemorrhage in young children should alert neurosurgeons to a possible diagnosis of atypical teratoid/rhabdoid tumor (AT/RT). Leptomeningeal dissemination is evident in 25 to 35% of cases at diagnosis. Gadolinium-enhanced MRI is more sensitive than CT in detecting small cortical and basal metastases (Fig. 28.3).
Controversial Point
• Intermittent surveillance imaging is frequently performed but is of no proven benefit.
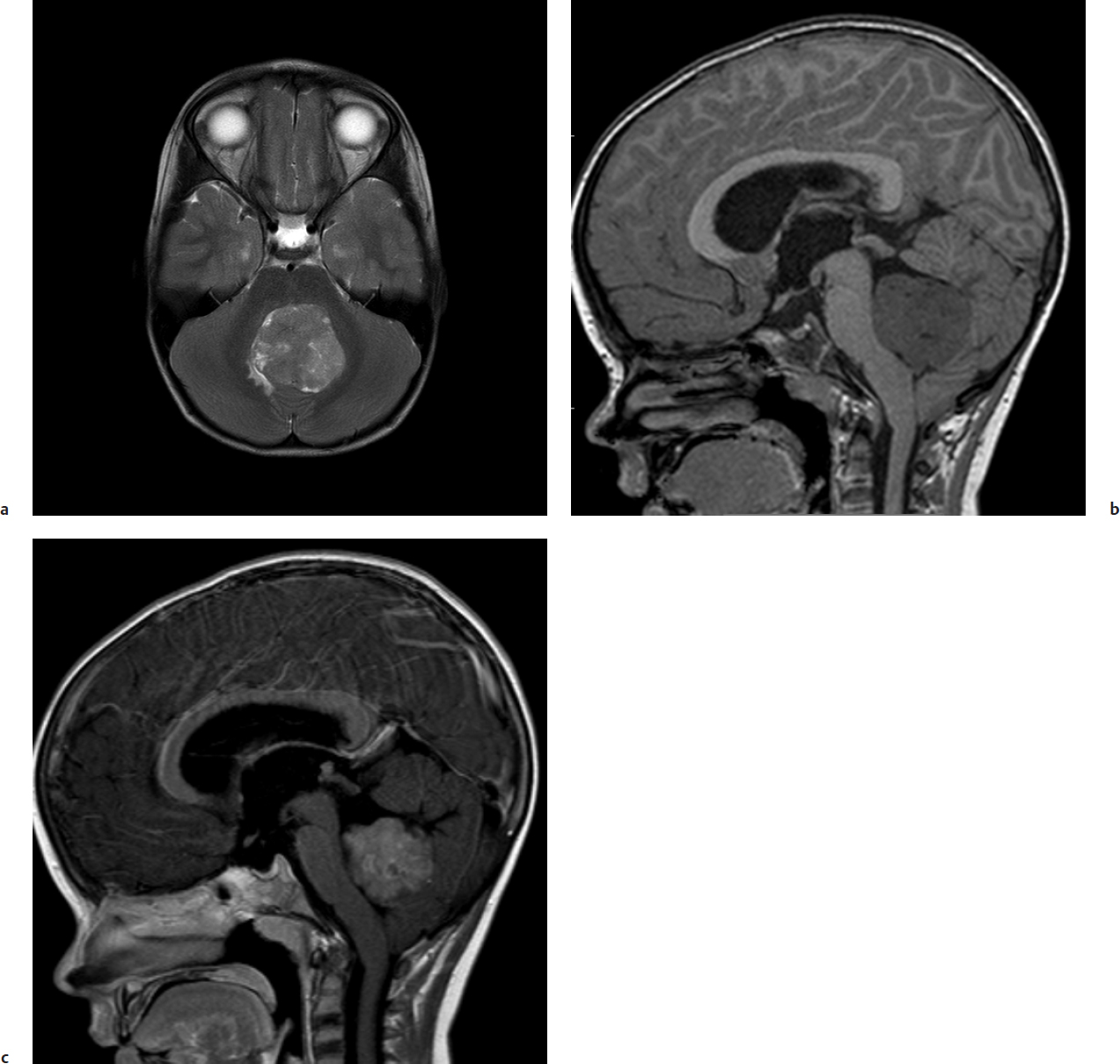
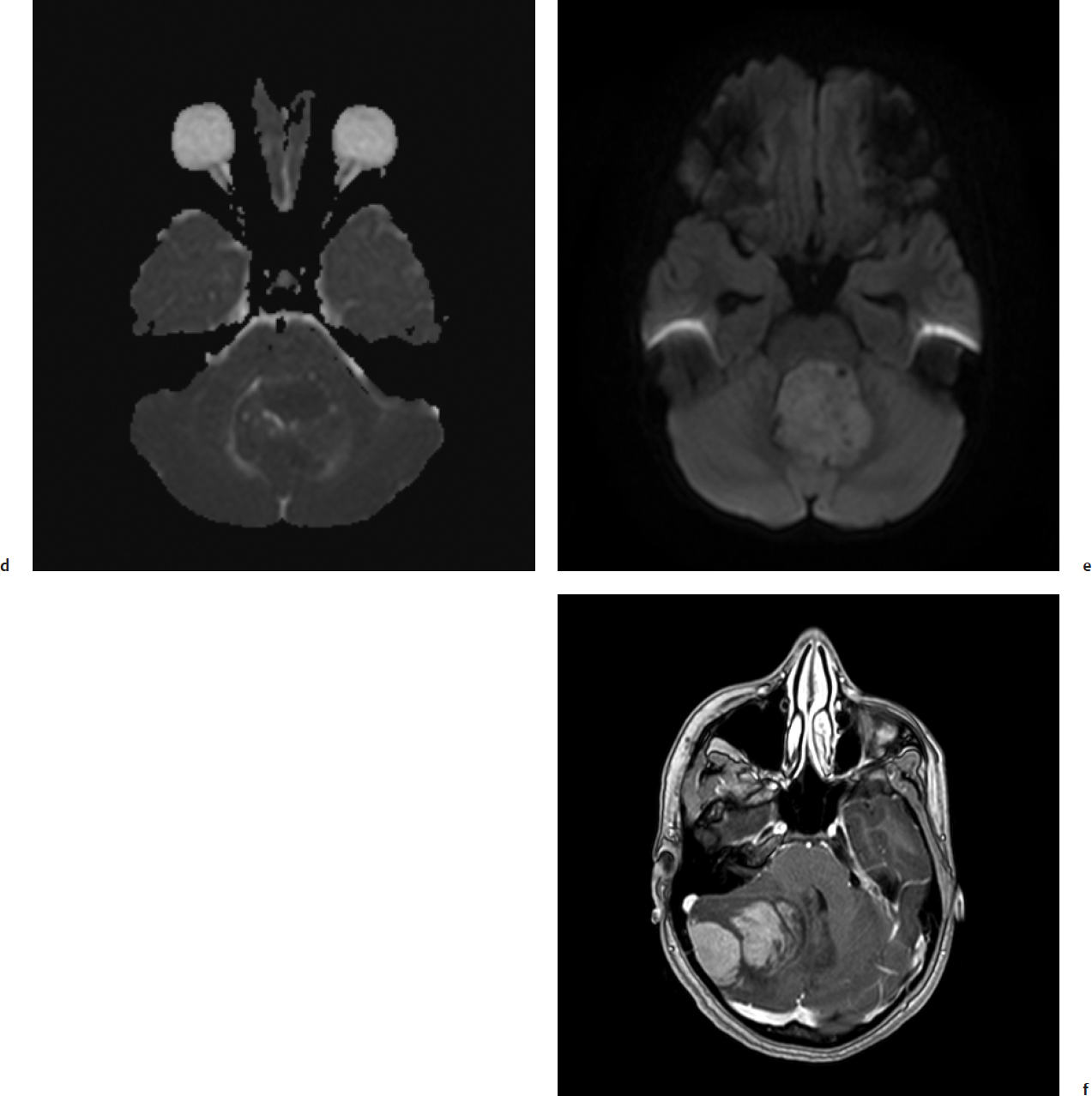
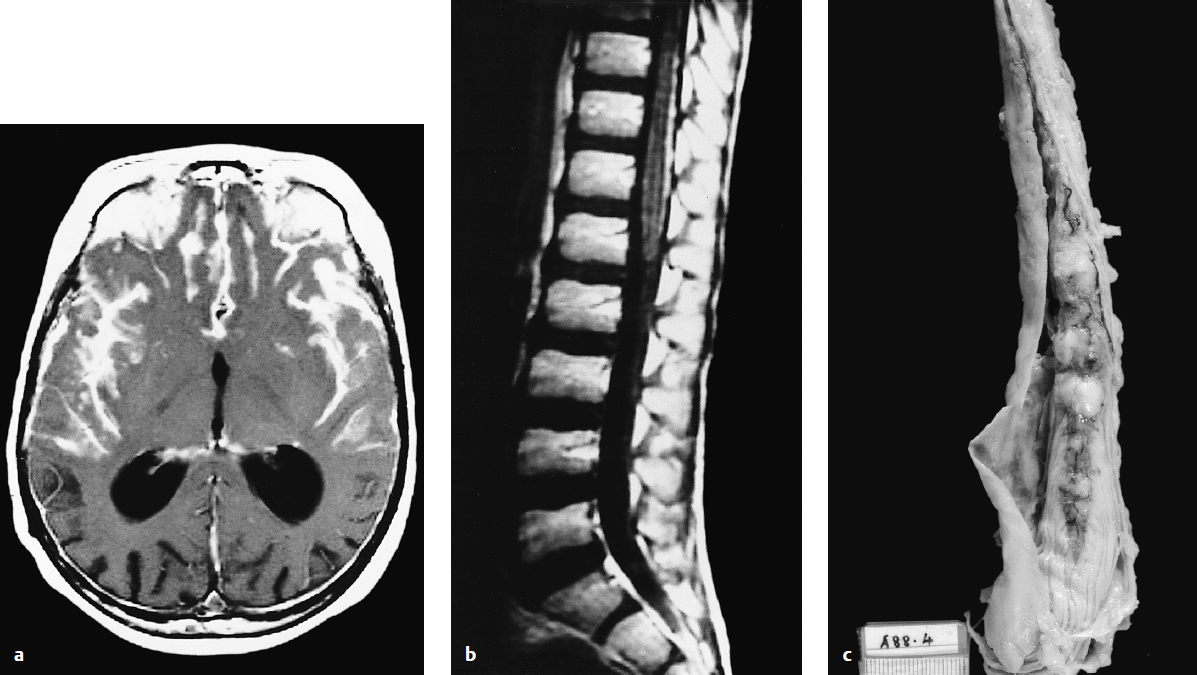
Surgical assessment of the extent of resection is often poor. Postoperative imaging should be performed within 48 hours of surgery when imaging artifacts due to postsurgical change are at a minimum.15
The most common site of intracranial recurrence is the primary site in the posterior fossa. Other common sites include the infundibular stalk, ventricular system, surface of the spinal cord, and the subfrontal subarachnoid space.
Pathology
Grossly, the surgeon finds a pinkish, gray mass that fills the fourth ventricle and often has tiny vessels around the periphery.1 The tumor typically arises from the medullary velum. CSF dissemination has occurred in 20 to 50% of cases at the time of operation, and on occasion can be seen as a whitish layer or “sugar coating” over the exposed cerebellum. Histopathological examination reveals an extremely cellular tumor with round cells, basophilic nuclei, a high nuclear to cytoplasmic ratio, and frequent mitoses.
Five histological variants are recognized by the 2007 World Health Organization (WHO) classification16: (1) Classic medulloblastomas (70 to 80% of cases) have densely packed and primarily undifferentiated cells with highly hyperchromatic nuclei surrounded by scanty cytoplasm and numerous Homer-Wright rosettes (tumor cell nuclei arranged in a circle around tangled cytoplasmic processes) (Fig. 28.4). (2) Desmoplastic tumors (15% of cases) occasionally show maturation to more differentiated ganglion cell tumors, and a biphasic pattern with distinctive lucent areas referred to as “pale islands” and areas with abundant internodular reticulin. The desmoplastic variant is more likely than the classic medulloblastoma to occur laterally in the cerebellar hemisphere and may have a better prognosis. Immunohistochemistry of medulloblastoma may show glial, neuronal, and ependymal differentiation. (3) Medulloblastomas with extensive nodularity (MBEN) consist almost entirely of nodules with large areas of neuropil and little or no desmoplastic internodular tissue. This variant is rare and shows extensive neuronal differentiation, has a very good prognosis, and occurs in very young children.17,18 (4) Anaplastic tumors have pleomorphic, enlarged nuclei and an increased cytoplasm-to-nucleus ratio and brisk mitotic activity. The nuclei display angulation and molding. (5) Large-cell medulloblastomas are composed of large cells with round nuclei and prominent nucleoli. Characteristic features include high mitotic rate, extensive apoptosis, and wrapping of tumor cells over each other.
Controversial Point
• Some authorities liken medulloblastoma to pineoblastoma and supratentorial primitive neuroectodermal tumor (PNET), all of which have similar histopathological appearance. These authorities group all of these tumors together under the rubric of PNET. More recent biological studies show that these tumors are in fact distinct entities with different clinical and biological profiles, and that they should not be lumped together.
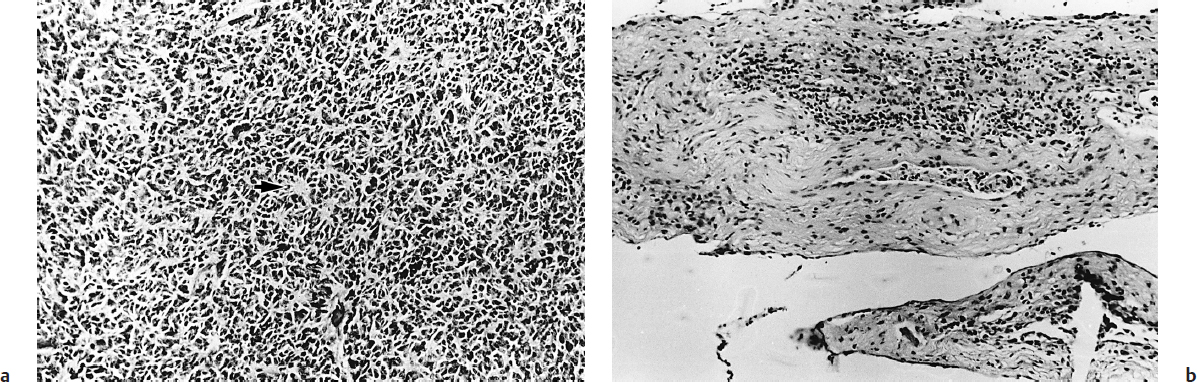
Atypical teratoid/rhabdoid tumor can also occur in the posterior fossa, and under the microscope some areas may contain only sheets of small blue cells, making it difficult to differentiate from medulloblastoma. The diagnosis of AT/RT is made by the presence of rhabdoid cells, which are medium-sized cells with an eccentric nucleus and a pink cytoplasmic inclusion body.19 The great majority of AT/RTs have mutations of the hSNF5/INI1 gene on chromosome 22, encoding for a protein that facilitates the transcriptional activation of genes by altering the structure of chromatin.20 Indeed, because histological identification of an AT/RT can be difficult, current Children’s Oncology Group (COG) protocols that include infants with AT/RT require that the hSNF5/INI1 gene be analyzed, as well as looking for deletions on chromosome 22. In addition, loss of INI1 expression is a very sensitive and specific molecular characteristic of AT/RTs.16 Patients with AT/RTs have a much worse prognosis than patients with medulloblastoma, despite recent improvements in treatment success with intensive multimodality treatment.21
Cerebrospinal Fluid Diversion
Many, if not most, children with medulloblastoma present with hydrocephalus. In the past many surgeons placed preoperative shunts or external ventricular drains in these patients. This was thought to immediately decrease the intracranial pressure; to enable the child to recover emotionally, physically, and nutritionally before the resection; to give the parents time to adjust to the diagnosis; and to allow resective surgery to be done on an elective basis by the best neurosurgical operating team. More recently, some groups are treating all children with posterior fossa tumors with a pre-resection endoscopic third ventriculostomy.22 Currently preoperative CSF diversion surgery is not recommended; most patients can be managed with corticosteroids followed by semiurgent tumor resection. Not all patients require a CSF diversionary procedure once the tumor is removed, and preoperative shunting or third ventriculostomy may expose these children to unnecessary risks.
About 25% of patients with medulloblastoma ultimately develop postoperative hydrocephalus requiring definitive treatment. The clinical presentation of postoperative hydrocephalus is that of increased intracranial pressure, or it may take the form of a pseudomeningocele at the site of the surgical incision. Many pseudomeningoceles resolve with serial lumbar punctures, but others may require VP shunting or cyst-peritoneal shunting. The risk of subsequent hydrocephalus and the need for a shunt is typically higher in younger patients and in the setting of metastatic disease, aseptic meningitis, and incomplete resection.
Controversial Point
• In most patients with medulloblastoma, a preoperative ventriculoperitoneal (VP) shunt, third ventriculostomy, or external ventricular drain is unnecessary and undesirable because symptomatic patients can usually be managed with corticosteroids alone until posterior fossa surgery is performed.
• Patients with high intracranial pressure and a large posterior fossa mass who undergo rapid drainage of CSF by VP shunt or external ventricular drain are at risk of clinical deterioration from intratumoral hemorrhage.
Treatment
Surgery
Surgical goals in the treatment of posterior fossa medulloblastoma include establishing a tissue diagnosis, reconstitution of CSF pathways, and removal of as much of the tumor as can be done safely. The tumor is approached through a midline suboccipital incision and can often be seen as a mass hanging down between the tonsils after the dura is opened. The surgeon should inspect the surface of the exposed cerebellum and spinal cord for evidence of leptomeningeal spread of disease. Gross total resection of disease is the surgeon’s aim; however, in about one third of cases the tumor will have invaded the brainstem. The tumor should not be “chased” into the brainstem because this will lead to serious neurologic morbidity.
The mortality rate for posterior fossa tumor surgery is low, but there is considerable morbidity secondary to damage to nearby structures such as the cerebellum, brainstem, and cranial nerves. Mutism is observed in over 20% of patients,23 and has been pathophysiologically linked with splaying of the middle cerebellar peduncles.24 The patient’s speech usually recovers in a few weeks to months after a brief period of dysarthria, but may not recover completely in some cases.25
Controversy
• Some authors believe that any residual tumor seen on postoperative imaging is a poor risk factor, but others feel that minimal residual disease (< 1.5 cm3) will not affect outcome. The role of second-look surgery to resect small residual medulloblastomas is controversial, especially in the setting of metastatic disease.
Radiotherapy
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


