Chapter Outline
Clinical Presentation and Risk Factors
Central Nervous System Involvement
Minimal Residual Disease Monitoring
DOWN SYNDROME WITH TRANSIENT MYELOPROLIFERATIVE DISORDER AND ACUTE MYELOGENOUS LEUKEMIA
Acute Myelogenous Leukemia
Acute myelogenous leukemia (AML) comprises a heterogeneous group of disorders characterized by the malignant clonal transformation of a hematopoietic stem or progenitor cell. In recent years, deeper understanding has been gained into the chromosomal changes and specific genetic mutations that precipitate this transformation. AML is a relatively rare disorder in children, with between 500 and 600 newly diagnosed patients in the United States each year. Because of the heterogeneous nature of AML, it has proven difficult to find a treatment strategy that is effective for all patients; thus, overall cure rates for AML have improved only modestly in the past few decades, lagging behind those for acute lymphoblastic leukemia (ALL). Drawing from data from the Medical Research Council (MRC) trials, patients with AML are now risk stratified based on cytogenetic features and emerging data on the prognostic significance of minimal residual disease (MRD) early in therapy. Current ongoing clinical trials are attempting to minimize toxicity for patients with low-risk disease and intensify therapy for patients with high-risk disease. As the molecular genetics behind the biology of AML are better elucidated, there is a pressing need to develop new strategies for targeted therapy to improve outcomes in AML.
Epidemiology and Etiology
According to the National Cancer Institute (NCI) Surveillance Epidemiology and End Results (SEER) data, it was estimated that each year, from 2006 to 2010, 5.1 per 100,000 children younger than 14 years and 4.6 per 100,000 adolescents aged 15 to 19 years were diagnosed with cancer. AML is less common than ALL: it comprises about 16% of childhood leukemias in children younger than 15 years but 36% of leukemias in adolescents 15 to 20 years, for a total of 500 to 600 new pediatric AML patients annually in the United States. AML has a bimodal distribution, occurring with higher incidence in children younger than 2 years and then again in adolescents 15 to 20 years old.
In ALL, boys are more commonly affected than girls; the incidence of AML is similar for all pediatric age groups, however, regardless of gender. Similarly, although the incidence of childhood ALL is higher in white than in black children, the incidence of AML is comparable for both races across all pediatric age groups. With regard to ethnicity, there is a higher incidence of AML in Hispanic versus non-Hispanic children. Of interest, although the incidence of ALL for children younger than 15 years increased from 1977 to 1995, the incidence of AML did not change significantly over this time period.
For a subset of patients with AML, a series of risk factors predisposing to the development of AML has been identified ( Box 51-1 ) These risk factors fall into two general categories: exposures (environmental or toxic) and genetic predisposition. The best understood of the exposures is prior treatment with chemotherapy and radiation. Alkylating agents (e.g., nitrogen mustard, cyclophosphamide, ifosfamide, chlorambucil, and melphalan) are associated with the development of myelodysplastic syndrome (MDS) and secondary AML, with deletions of chromosomes 5 and 7. Treatment with topoisomerase II inhibitors such as the epipodophyllotoxins has been linked to a specific type of AML with chromosome band 11q23 translocations, which produce fusion proteins involving the mixed-lineage leukemia (MLL) gene (MLL) . Treatment with anthracyclines in adults with breast cancer, or in children for leukemias and sarcomas such as osteosarcoma, is associated with the development of secondary leukemias, including ALL, AML, and chronic myelogenous leukemia (CML). Although therapy-related AML generally carries a worse prognosis than de novo AML, there have been reported cases of therapy-related AML harboring the better risk cytogenetic abnormalities: t(8;21), inv(16), t(16;16), and t(15;17), often in conjunction with other cytogenetic abnormalities. In the case of therapy-related AML with inv(16) and t(16;16), a rare core-binding factor β (CBFβ) –myosin heavy chain 11 (MYH11) fusion transcript has been reported, with different breakpoints compared with the usual fusion transcript found in better-risk de novo AML. Similarly, there is a case report of a patient with therapy-related AML with t(8;21), and the breakpoint at 21q22 was outside the AML1 locus.
Prenatal Exposures
Alcohol
Pesticides *
* Conflicting or limited data.
Foods naturally high in topoisomerase II inhibitors *
Viral infections
Environmental Exposures
Ionizing radiation
Chemotherapeutic agents
Alkylating agents
Epipodophyllotoxins
Anthracyclines
Organic solvents (e.g., benzene)
Radon *
Pesticides *
Viral infections *
Hereditary Conditions
Down syndrome
Noonan syndrome
Neurofibromatosis
Fanconi anemia
Bloom syndrome
Severe congenital neutropenia (Kostmann syndrome)
Shwachman-Diamond syndrome
Klinefelter syndrome (XXY) *
Germline Predisposition Mutations
NF1 (for JMML)
RUNX1
CEBPA
GATA2
ANKRD26
Acquired Disorders
Aplastic anemia
Paroxysmal nocturnal hemoglobinuria
JMML, Juvenile myelomonocytic leukemia.
Radiation exposure may be acquired from the environment or through medical procedures and is well documented to predispose to leukemia. The increased risk for AML and CML in survivors of nuclear blasts in Japan who were exposed to excessive doses of ionizing radiation has been well documented, and these patients have a higher rate of abnormalities of chromosomes 5 and 7. An increased risk for AML and ALL in children exposed to radiation in utero has been described, particularly in the past, when diagnostic radiographs were more common during pregnancy, although a retrospective study of children with leukemia in Sweden did not confirm this finding. Nuclear power plant workers have a higher risk for leukemia, although there does not appear to be a higher risk in residents living near such plants. Air travel is a source of exposure to cosmic radiation, and studies have shown an increase in malignant melanoma and other skin cancers in flight crew members, an increase in breast cancer in female flight crew members, and an increase in AML in male cockpit crew members who logged more than 5000 flight hours annually. Studies designed to measure the potential risk for malignancy from residential or occupational exposure to magnetic fields around high-voltage electrical lines have generated conflicting results, and a causal relationship with AML has not been conclusively established.
There are several other environmental exposures with possible links to AML, some of which have yet to be proven definitively. Benzene exposure, which may occur occupationally or through tobacco smoking, has been shown to increase the risk for AML. Several prenatal exposures have been studied as risk factors for the development of AML, particularly in children younger than 3 years. Maternal alcohol consumption during pregnancy was studied in a Children’s Cancer Group (CCG) case-control study and found to have an association with increased rates of childhood AML in a dose-dependent matter, with an odds ratio of 2.64 overall and an odds ratio of 7.62 for classic French-American-British (FAB) classification category M1 (myeloblastic with minimal maturation) and M2 (myeloblastic with maturation) AML; other case-control studies have confirmed this finding. Some early studies had suggested an association between maternal tobacco and marijuana use and childhood AML, but more recent analyses have not confirmed this association. A Children’s Oncology Group (COG) case-control study looked at maternal consumption of foods such as soy, green and black tea, cocoa, red wine, and certain fruits and vegetables that are naturally high in DNA topoisomerase II inhibitors and found an odds ratio of 1.9 to 3.2 for the development of MLL -rearranged AML in the setting of high maternal consumption of these foods. Some studies have suggested an increased risk for childhood AML after high exposure to pesticides, either prenatally or postnatally, up to 3 years of age. There is interest in an association between viral infections and AML, but few data. One case-control study has suggested an increase in childhood AML in offspring of mothers who reactivated Epstein-Barr virus during pregnancy. Parvovirus B19 is associated with pure red cell aplasia, and certain human leukocyte antigen (HLA)-DRB1 alleles seem to be associated with symptomatic infection. One study of 16 leukemia patients suggested an association between parvovirus B19 infection and acute leukemia in 4 of the patients, including 1 patient with AML, all of whom carried these particular HLA-DRB1 alleles, although this association needs further investigation. Several retrospective studies have suggested that breastfeeding may be protective against ALL and AML. Risk related to indoor radon exposure is controversial. One French study showed a higher rate of AML in children with high exposure but other studies have showed no association. Rarely used now, chloramphenicol use in children was formerly associated with an increased risk for AML.
An increased incidence of AML has been observed for patients with certain hereditary disorders. Children with congenital disorders of myelopoiesis, such as Kostmann syndrome, Shwachman-Diamond syndrome, and Diamond-Blackfan anemia, are predisposed to AML. Patients with inherited syndromes associated with chromosome fragility and impaired DNA repair mechanisms, such as Fanconi anemia and Bloom syndrome, also have an increased risk for development of AML. Neurofibromatosis type 1, which is caused by mutations in the neurofibromin tumor suppressor gene on chromosome 17, is associated with juvenile myelomonocytic leukemia (JMML). Patients with certain constitutional chromosomal abnormalities carry a higher risk for AML. Down syndrome is one of the most clinically prominent examples in this category. It is estimated that over 10% of infants with Down syndrome exhibit a transient myeloproliferative disease associated with a GATA1 mutation. Down syndrome patients have a 10 to 20 times higher than average risk for acute leukemia. In Down syndrome patients younger than 4 years, AML, usually acute megakaryoblastic (AMKL), is far more common than ALL: an estimated 1 in 500 Down syndrome patients develop AML. In contrast to AMKL in the general pediatric population, which carries an extremely poor prognosis, this disease in Down syndrome patients is extremely sensitive to chemotherapy (see later). Although early studies did not suggest a higher risk for AML in patients with Klinefelter syndrome (XXY), more recent data suggest that these patients may have a higher risk for hematologic malignancy, including AML. More recently, a number of MDS/AML predisposition genes have been identified, including loss of function alterations affecting the key hematopoietic transcription factor genes RUNX1, CEBPA , and most recently GATA2 .
Although the predisposing risk factors and genetic conditions described may provide insight into the causative mechanisms that increase the risk for development of AML, most patients with de novo AML have no known predisposing exposures or conditions. Point mutations and chromosomal deletions and translocations occur at a background rate, even in healthy individuals, during hematopoietic stem and progenitor cell expansion. The extent to which inherited, expressed, single-nucleotide polymorphisms in the general population alter mutational rates during myelopoiesis and increase the risk for AML and other cancers remains an area of intense investigation.
Biology
Clonal Origin of Myeloid Leukemia Cells
Normal myelopoiesis is a complex differentiation program whereby primitive hematopoietic stem cells (HSCs) develop along a multistep pathway into fully differentiated, functionally active circulating blood cells. This exquisitely controlled process is regulated by the intricate interactions among the expression levels of various transcription factors, growth factors and their receptors, cytokines, enzymes, and still unidentified novel molecules. A series of sequential genetic abnormalities perturbs this normal developmental progression and leads to AML. Although the relationships between specific morphologic subtypes of AML and their specific recurring genetic abnormalities have provided some insight into the mechanisms of leukemogenesis, understanding of the process whereby these fusion gene products interact with normal signal transduction pathways to subvert hematopoietic cell development is incomplete.
Several strong lines of evidence support the hypothesis that AML progresses from a single transformed hematopoietic stem or progenitor cell. More than 30 years ago, studies were pioneered by Beutler and colleagues and Fialkow using X inactivation patterns in female patients to establish the clonal origin of human malignancies, including leukemia. By showing the presence of a single glucose-6-phosphate dehydrogenase (G6PD) isoenzyme in the leukemic myeloblasts of heterozygous females, the clonal origin of myeloid leukemia cells was demonstrated. Females who are heterozygous at the G6PD locus express two isoforms of the enzyme. Approximately half of the cells in normal somatic tissue have randomly inactivated one of the X chromosomes, and approximately half of the cells should therefore express each isoform. Unlike normal somatic cells, AML cells of female patients heterozygous for G6PD expressed only one G6PD isoform, indicating cells of clonal origin. In the 1980s, Vogelstein and coworkers developed a strategy using X chromosome–linked DNA restriction length fragment polymorphisms to determine the clonal origin of human tumors. They established that maturing granulocytic cells arise from the malignant clone in patients with AML. Later, X chromosome inactivation to determine clonality in malignancies was carried out using the polymerase chain reaction (PCR) assay to distinguish X-linked polymorphic genes.
Transformation of Stem Cells with Self-Renewal Capacity
These earlier studies, and those performed by Bonnet and Dick and associates in the 1990s, led to the insight that leukemia cell populations form a hierarchy in which leukemia stem cells or leukemia-initiating cells (LICs) are responsible for their own self-renewal and for the generation of the more differentiated progeny within the leukemic clone. This theory was in contrast to the stochastic model, which stated that malignant properties within cancer cell populations follow Gaussian distributions, resulting in the random or stochastic probability that individual cancer cells within the population are able to initiate malignant progression. Whereas AML was the first human cancer in which cancer stem cells were identified, the case has now been made in breast cancer, brain tumors, and colon cancer, among others. In general, AML stem cells are believed to arise through the transformation of HSCs, which then retain the capacity for self-renewal, or through transformation of more differentiated hematopoietic progenitor cells that acquire the ability to self-renew by virtue of the particular transforming mutations, such as the formation of an MLL fusion gene.
AML is a heterogeneous disease in its clinical manifestations, response to therapy, and molecular genetics; thus insight into the cell of origin would have important ramifications regarding diagnosis and treatment. Moreover, refractory disease and relapse are often attributed to the presence of an LIC population that is resistant to chemotherapy; thus the ability to identify and target this subpopulation may be key to improving overall disease outcome. In the 1990s, Dick’s group produced a series of convincing papers that strongly implicated the primitive, pluripotent HSC as the LIC in some types of AML. “Stemness,” or the capacity to function as an LIC, includes self-renewal, proliferation, and differentiation and is tested in vitro by replating limiting dilution studies and in vivo by serial transplantation assays with the ability to regenerate a clonal leukemia in secondary and subsequent recipient animals. The HSC, with its inherent gene programs ensuring self-renewal, proliferation, and survival appears to be an ideal target to become sabotaged through a series of genetic events, leading ultimately to a fully transformed AML LIC that is capable of producing a clonal population of leukemia cells. According to this argument, different AML phenotypes often occur as a result of the gene(s) that are altered, leading to differentiation arrest within the progeny of the LIC, but not necessarily reflecting the degree of the commitment of the initially transformed cell. Support for this hypothesis initially came from several clonality studies in leukemic cells from patients with AML, which demonstrated multiple-lineage involvement in a high proportion of cases, and implicated a multipotent HSC as the cell of origin. More supportive evidence has come from studies looking at cytogenetic markers and characteristic cell surface antigen expression patterns. Pluripotent HSCs express CD34 but do not express CD38 or HLA-DR, whereas more committed myeloid progenitor cells are CD34+, CD38+, and HLA-DR+. Using fluorescence-activated cell sorting (FACS) of leukemic cell populations, the same two subpopulations, CD34+/CD38− and CD34+/CD38+, were isolated from the bone marrow of patients with different AML subtypes. The two subpopulations were then evaluated using fluorescence in situ hybridization (FISH) to determine the presence of cytogenetic abnormalities. The studies detected the same characteristic cytogenetic abnormalities in the CD34+/CD38− stem cell fraction as in the original leukemic bone marrow samples, implying origin in a very early HSC compartment.
Further evidence supporting the stem cell origin model of AML was derived from transplantation experiments in which purified human AML cells were transplanted into mice with severe combined immunodeficiency (SCID). These experiments defined a SCID mouse leukemia–initiating cell (SL-IC) in the bone marrow of patients with AML and showed that these SL-ICs are CD34+/CD38− and that their engraftment produces large numbers of colony-forming progenitors. The CD34+/CD38+ and CD34− fractions did not engraft. Similar results were obtained through transplantation studies in a modified SCID mouse, the nonobese diabetic mouse with SCID (NOD/SCID), with cells from patients with AML. These studies identified and analyzed AML stem cells from samples of different FAB subtypes on the basis of their ability to initiate human AML after transplantation in NOD/SCID mice. These SL-ICs were able to proliferate and differentiate after transplantation, producing disease in the mice identical to that in the donor, as well as being able to renew themselves, reestablishing AML in secondary recipients, demonstrating that SL-ICs are capable of self-renewal. Again, the SL-ICs were found to reside in the CD34+/CD38− fraction and not in the CD34+/CD38+ or CD34− fractions. The SL-IC phenotype was consistent regardless of the FAB subtype (e.g., M1, M2, M4, M5). As few as 2 • 10 4 CD34+ cells were able to initiate the leukemic clone in recipient mice, whereas 100 times as many CD34− cells failed to engraft. Cells with the CD34 surface antigen were further fractionated on the basis of CD38 expression. Only the CD34+/CD38− fraction contained SL-ICs.
Whereas the transformation of the HSC provides an attractive mechanism to generate hierarchical myeloid leukemia cell populations, several studies have given new credence to a second mechanism through which committed progenitor cells along the pathway of myeloid differentiation are vulnerable to transforming mutations that confer self-renewal properties to progenitor cells that inherently lack this ability. These transformation events create an LIC capable of generating a population of aberrant cells blocked at the differentiation state at which the initial transforming event occurred. Several translocations involving the MLL gene have been shown to initiate self-renewal in myeloid progenitor cells and to result in an AML phenotype. A head-to-head comparison was conducted by retrovirally transducing the human AML-associated MLL-ENL fusion gene into murine HSC, common myeloid progenitors (CMPs), and granulocytic-monocytic–restricted progenitors (GMPs) and evaluating the relative transforming ability and resultant phenotype in each cell type. AML developed in mice transplanted with any of the three transduced cell populations and, in each case, displayed an identical myelomonocytic phenotype as assessed by morphology and flow cytometry. Similarly, the MOZ-TIF2 fusion gene was able to confer self-renewal properties to committed myeloid progenitor cells and resulted in AML when transplanted into irradiated mice. Although the fusion genes MLL-ENL and MOZ-TIF2 appear to impart leukemogenic capabilities to committed progenitor cells convincingly, other fusion genes such as BCR-ABL can induce leukemic transformation only when expressed in an HSC but not a more mature myeloid cell. Similarly, the potent oncogenic combination of Hoxa9 and Meis1 only resulted in AML when transduced into HSCs but not in more committed myeloid progenitors. In this case, expression of β-catenin was found to be the necessary factor for oncogenesis. Present in HSCs but absent in more committed progenitors, the addition of β-catenin to progenitors transduced with Hoxa9 and Meis1 was sufficient to lead to AML. These findings point to the differing transforming abilities of different oncogenes that are likely cell-type and cell-context specific. Hence both cell-of-origin hypotheses appear to be correct, depending on the specific oncogene or tumor suppressor that provides the initiating genetic lesion. Increased proliferation, survival, and self-renewal are all necessary attributes of a leukemia stem cell. Some genetic abnormalities, such as MLL-ENL and MOZ-TIF2 , may be capable of inducing self-renewal and thus are able to transform a more differentiated myeloid cell into an LIC. By contrast, other oncogene fusions, such as BCR-ABL , or oncogenic transcription factor genes, such as Hoxa9 , do not possess self-renewal capabilities and thus require a cell that inherently has this characteristic, namely, the HSC.
More recently, challenges to the cancer stem cell hypothesis have arisen out of a number of studies, predominantly in solid tumors such as melanoma, where the frequency of cells capable of giving rise to a new tumor in an immunocompromised host is of sufficient frequency to call into question whether the LIC represents a unique subpopulation or the original stochastic model of cancer progression holds true. These studies have again called into question previously accepted tenets. CD34+/CD38+ populations, in addition to the CD34+/CD38−, have also been found to contain LICs when injected into more immune-deficient hosts that lack key components of the innate immune system. These studies and additional syngeneic and cogenic transplant studies in mice as well as zebrafish highlight the critical context of the recipient animal in determining the LIC. The heterogeneity of LICs in AML is further demonstrated by an increasing number of cell surface markers in addition to CD34 and CD38, including CD123, TIM3, and CD47. Additionally, a number of studies have attempted to identify a specific LIC genetic signature, which has been found at least in some cases to independently predict prognosis. In this case, the LIC signature was reminiscent of the normal HSC, hearkening back to the original predictions of Dick’s group highlighting the HSC as a putative LIC cell of origin. However, although these findings are provocative, a consistent cadre of LIC genes has not yet been established. Clonality studies demonstrating the progressive acquisition of mutations in a given cell have also been put forward as an alternative to the cancer stem cell hypothesis. Although seemingly at odds, these different concepts can be reconciled into a single integrated framework. Myeloid (and lymphoid) leukemia-initiating events may occur in both HSCs and committed progenitor cells, and each leukemia arises from a combination of multiple genetic abnormalities. Secondary events subsequently are acquired in different subpopulations, generating unique clones with varying malignant potential depending on the genetic lesion and the self-renewal capacity of the cell in which these abnormalities occur. The number of genetic lesions necessary to cause frank AML is being defined based on next-generation sequencing studies recently published by the group at Washington University in St. Louis as well as other centers. The evolution of particular clones appears to determine the eventual phenotype of the AML cells documented at diagnosis ( Fig. 51-1 ).
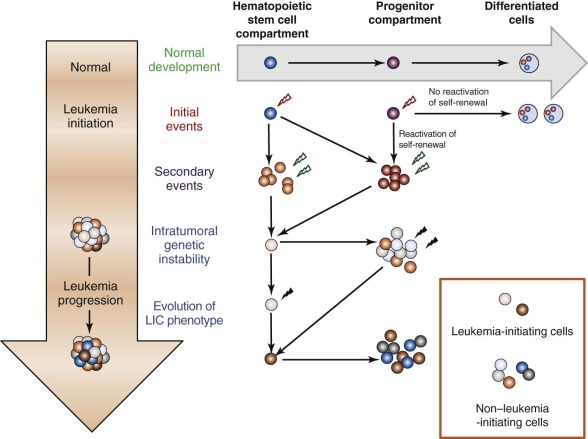
These new discoveries have added additional complexity to our understanding of the contribution of LICs to AML pathogenesis by demonstrating that the heterogeneity inherent in this disease extends to the AML stem cells. However, the identification of new LIC surface markers, genetic signatures, and gene-specific mutations hold out promise that LIC-targeted therapies will be realized in the coming years.
Molecular Genetics
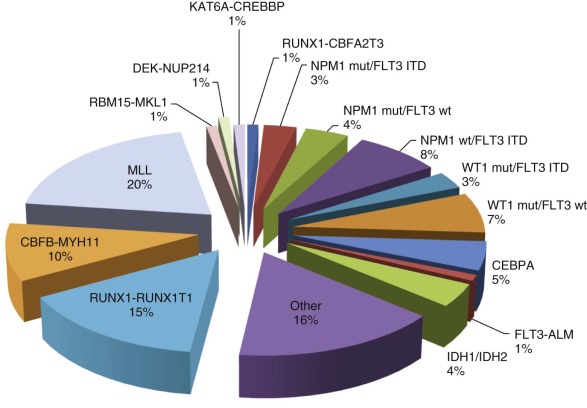
Recent AML protocols have incorporated current knowledge regarding the significance of specific genetic abnormalities to direct risk-stratified approaches to treatment. Chromosomal abnormalities and, in particular, translocations identified on cytogenetic examination of the blast cells represent the initial genetic lesions incorporated into disease taxonomy and formed the basis of the World Health Organization (WHO) classification in 2008. Careful analysis of these translocations and other recurring genetic abnormalities in leukemia patients has had a profound impact on our understanding of the molecular genetic basis of leukemia ( Fig. 51-2 ). Although many chromosomal abnormalities from AML blast cells have been identified and studied extensively, no single mutation has been shown to be sufficient to cause acute leukemia. These findings prompted Gilliland and colleagues to propose what is now considered the classic “two-hit model” of AML pathogenesis. In this model, leukemic transformation results from distinct but collaborative sequential mutations in parallel molecular pathways that affect cell survival, proliferation, differentiation, and self-renewal. Class I mutations frequently involve an activated receptor or cytoplasmic-nuclear tyrosine kinase conferring a proliferative and/or survival ability to a specific cell but not affecting differentiation. By contrast, class II mutations specifically result in differentiation arrest and/or self-renewal and often involve key transcription factor oncogenes but alone do not confer a proliferative advantage. Although attractive, this model likely represents an oversimplification of AML pathogenesis. Initially, microarray studies and more recently, deep-sequencing approaches have revealed the presence of novel mutations and absence of tyrosine kinase lesions, particularly in normal karyotype AML, calling into question the general applicability of the “two-hit” model in all cases of AML. However, although the category of genetic lesions may not always subscribe to this classic paradigm, the number of “hits” required may be more universal. Genomic profiling of individual patient leukemias suggest that in some cases only two to three lesions are required for clonal evolution to frank AML, with fewer “hits” necessary in pediatric AML than in its adult counterpart. Regardless, the prevalence of lesions categorized by the “two-hit” model will be outlined here, followed by more recent discoveries of novel genes implicated in AML pathogenesis and the variable frequency of these lesions in pediatric versus adult AML.
Class I Mutations
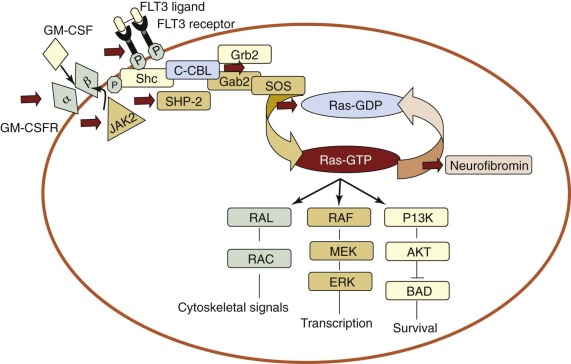
Among the earliest examples of class I mutations, RAS mutations were described in the early 1980s. There are three functional RAS gene family members— NRAS, KRAS, and HRAS —that all act as guanosine nucleotide phosphate (GTP)-binding proteins. Mutations of NRAS are the most prominent RAS mutations in AML and have been found in as many as 30% of adult patients with AML. Mutations, most frequently in codons 12, 13, and 61, result in the retention of the RAS protein in its active GTP-bound state and lead to the constitutive activation of downstream effector proteins, which causes the transcriptional activation of various target genes that direct cellular differentiation, proliferation, and survival ( Fig. 51-3 ). RAS proteins may be alternatively activated by mutations in genes encoding a number of regulatory factors. Activating point mutations in the SHP-2/PTPN11 phosphatase provide a stimulatory signal through guanine nucleotide exchange factors, such as “Son of Sevenless” (SOS), resulting in increased RAS pathway signaling, as do inactivating mutations of neurofibromin-1 (NF1) , which codes for a GTPase-activating protein (GAP) of the same name. NF1, normally functions, at least in part, by increasing the GTPase activity of RAS, thereby inactivating RAS-GTP by converting it into the inactive RAS-GDP form. These three types of mutations function independently to activate RAS signaling and lead to disturbances in hematopoietic cell differentiation and proliferation that may ultimately contribute to the development of MDS, myeloproliferative disease (MPD), and AML (see Fig. 51-3 ). Mutations in NF1 and PTPN11 have been particularly associated with JMML (see later).
There has been significant disagreement regarding the prognostic implications of RAS mutations in MDS and AML. Several studies have demonstrated that RAS mutations in patients with MDS confer a poor prognosis and increase the risk for progression to acute leukemia. Other studies have reported no difference in survival in AML patients who carried RAS mutations compared with patients without RAS mutations, whereas some have suggested improved survival in patients with AML and NRAS mutations. A large study of more than 2500 patients with AML demonstrated no difference in prognosis in patients with or without RAS mutations. This study also demonstrated an association between RAS mutations and a leukemia karyotype containing inv(16), a class II mutation resulting in the CBFβ-MYH11 fusion protein and altered function of the core-binding factor (CBF) transcriptional complex. A recent report from the COG examining 825 pediatric AML samples treated on two recent trials identified NRAS mutations in 10% of patients predominantly in codons 12 and 13, with no mutations in codon 61. Interestingly, most of these mutations were associated with good prognosis NPM1 mutations (see later) and not CBF alterations. However, multivariate analysis confirmed a lack of independent impact of NRAS mutations on prognosis.
Another group of class I mutations involves constitutive activation of receptor or cytoplasmic-nuclear tyrosine kinases. The BCR-ABL translocation that is the sine qua non of CML is an example of a class I mutation causing the activation of the ABL tyrosine kinase. Similarly, receptor tyrosine kinase mutations activate the human protooncogenes KIT and FLT3 in AML. The KIT protein is activated by the binding of its ligand stem cell factor and is critical for the development and growth of mast cells, melanocytes, HSCs, and the interstitial cells of Cajal. Mutations in KIT allow ligand-independent activation of KIT and confer factor-free growth and tumorigenicity in hematopoietic cell lines. Blasts from patients with AML express KIT on their cell surfaces in most cases. Deletional and insertional mutations of KIT have been identified in the blast cells of patients with AML, often in association with the CBF abnormalities inv(16) or t(8;21).
The FLT3 gene encodes a class III fms-like receptor tyrosine kinase expressed in early hematopoietic progenitors. FLT3 mutations occur in two varieties, internal tandem duplications (ITDs) in the juxtamembrane region and activation loop mutations (ALMs), primarily at codons 835 and 836. The FLT3 -ITD is one of the most frequent abnormalities in adult AML, documented in 25% to 30% of patients, and generally confers a poor prognosis. By contrast, ALMs occur in only 10% of adult patients and have not been associated with inferior survival rates. FLT3 -ITDs have been observed in 5% to 16% of pediatric AML cases and ALMs in 2% to 5% of cases. Paralleling the adult studies, FLT3 -ITDs, but not ALMs, were associated with a poor prognosis in children. FLT3 -ITDs result in the constitutive activation of FLT3 and cause interleukin-3–independent growth in Ba/F3 and 32D cells. This activation, in turn, results in the signaling of multiple downstream pathways, including the aforementioned RAS pathway, and the inhibition of caspase-mediated apoptosis through the stimulation of phosphatidylinositol 3′-kinase (PI3K) and AKT (see Fig. 51-3 ). However, mice transplanted with FLT3-transformed hematopoietic cells develop a myeloproliferative disorder characterized by splenomegaly and leukocytosis but they do not develop AML. FLT3 -ITDs often occur in conjunction with other mutations, including NPM1 (see later) and WT1 (see later).
Class II Mutations
In class II mutations, the molecular defect appears to be at the level of transcriptional activation, whereby transcriptional repressors cause a dominant negative inhibition of normal hematopoietic cell differentiation. AML can be subtyped based on the type of class II mutations in the blast cells of patients. The type 1 subset of class II mutations occurs in patients with de novo AML. These are typical chromosomal translocations resulting in chimeric oncoproteins that in some cases may cause the inhibition of differentiation. The type 2 subset of class II mutations usually are found in patients with AML arising after a prodrome of MDS and manifest more commonly in older adults or in patients who have received previous chemotherapy. Cytogenetic studies of the blast cell populations in these patients have complex karyotypes, lacking translocations but harboring deletions such as 5q−, monosomy 7, and 20q−.
Type 1 Mutations
Translocations Involving Core-Binding Factor Complex.
Several of the translocations that have been identified in adult and childhood de novo AML involve the CBF complex. The CBF complex is the most frequent target of chromosomal translocations in the human leukemias. The CBF regulatory complex consists of a DNA-binding subunit, RUNX1 (also called CBFα or AML1) and CBFβ, a subunit that does not bind DNA independently but heterodimerizes with RUNX1 or one of its closely related family members. Chromosomal translocations that modify the CBF complex in AML include the following: t(8;21)(q22;q22), which generates the AML1-ETO fusion protein, more recently referred to as RUNX1-RUNX1T1 ( Fig. 51-4 ); t(3;21)(q26;q22), which gives rise to AML1-EVI1; and inv(16)(p13;q22), which fuses the CBFβ to smooth muscle myosin heavy chain (SMMHC), resulting in the CBFB-MYH11 fusion gene. The AML1 gene was inactivated in the germline of mice and was shown to be essential for definitive hematopoiesis of all cell lineages. Homozygous animals die early in embryogenesis of central nervous system (CNS) hemorrhage, but they have normal morphogenesis and yolk sac hematopoiesis lineages. Inactivation of the CBFβ gene in the mouse has shown a similar phenotype in the homozygous null mice. These experiments have demonstrated that the AML1/CBFα complex is essential for normal hematopoiesis and that chromosomal rearrangements involving this complex may interfere with its regulatory function in ways that lead to disruption of cellular differentiation, and eventually malignant transformation.
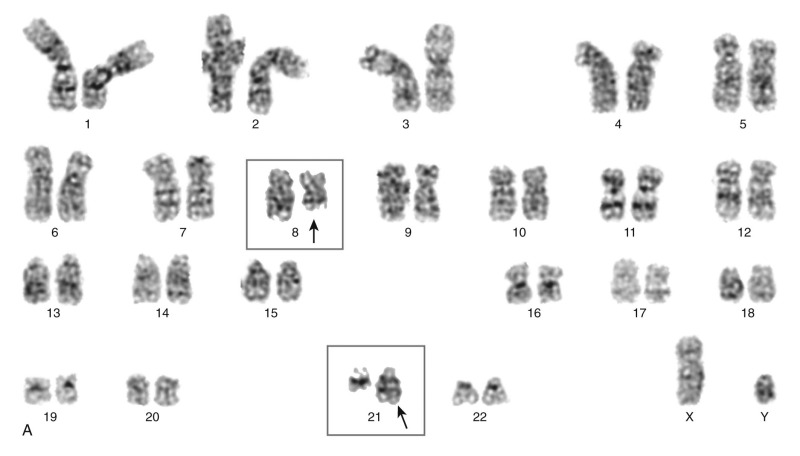
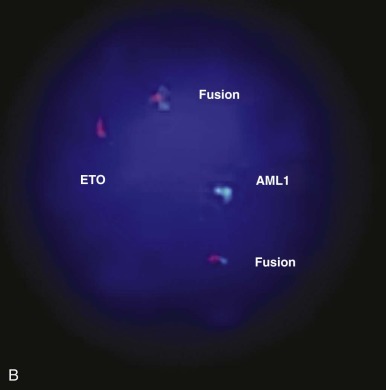
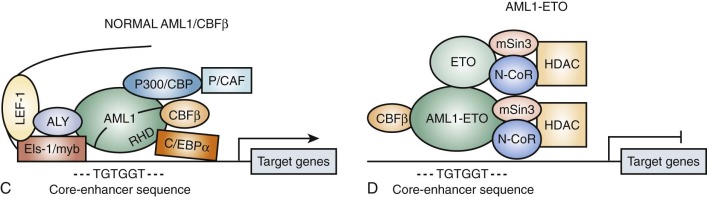
Evidence primarily from knock-in studies in mice has supported the hypothesis that the AML1-ETO fusion protein functions as a dominant negative inhibitor of wild-type functions. In these experiments, the AML1-ETO fusion gene is “knocked in” to the wild-type AML1 locus. The phenotype of these mice is identical to the AML1 knockout; the mice die early in embryogenesis from CNS hemorrhage and lack any evidence of definitive hematopoiesis, although the rare cells that survive and express AML1-ETO have an increased capacity for self-renewal. Biochemical experiments have shown that the ETO portion of the AML1-ETO chimeric protein recruits nuclear corepressor complexes to the CBF promoters, resulting in transcriptional inhibition of target genes normally activated by the AML1/CBFβ heterodimeric complex (see Fig. 51-4D ). Subsequent studies using inducible murine model systems to bypass the embryonic lethality of AML1-ETO overexpression have demonstrated that overexpression of this transgene at later developmental time points confers enhanced self-renewal capacity to bone marrow progenitors, as demonstrated by serial culture experiments. Importantly, the mice do not develop AML but appear to be predisposed to leukemogenesis. Thus after treatment with N -ethyl- N -nitrosourea (ENU), the mice developed rapid onset of AML compared with similarly exposed wild-type mice. Moreover, as noted, there is an association in myeloid leukemia of AML1-ETO translocations and mutant tyrosine kinases, such as KIT. Interestingly, AML1-ETO transcripts may persist for a long time in patients with AML1-ETO –expressing AML in sustained remission.
Translocations Involving the Retinoic Acid Receptor-α.
Reciprocal chromosomal translocations involving the retinoic acid receptor-α (RARA) gene locus on chromosome 17 are the defining molecular features of acute promyelocytic leukemia (APL). Before the RARA translocations were described, retinoic acid was known to induce myeloid differentiation in vitro and therapy with all- trans -retinoic acid (ATRA) in patients with APL was shown to induce complete remission (CR). The intense investigation of the RARA gene triggered by these observations culminated in identification of the promyelocytic leukemia PML-RARA translocation breakpoint, t(15;17)(q22;q11), reported simultaneously by several groups. Although most APL cases have the associated t(15;17), other reciprocal translocations involving the RARA gene have been identified in which the gene is fused to other gene partners. The most common of these include the promyelocytic leukemia zinc finger (PLZF) gene on chromosome 11 in t(11;17)(p13;q11) and the nucleophosmin gene (NPM) on chromosome 5 in t(5;17)(q31;q11).
Normally RARA functions as a ligand-dependent activator of transcription. It forms a heterodimer with the related protein, retinoid X receptor (RXR), and binds through its zinc finger domain to a specific promoter sequence; in the presence of the retinoic acid ligand, the RARA-RXR heterodimer activates transcription of retinoic acid–responsive genes. This process is accomplished in concert with a coactivator complex that includes proteins such as p300 and pCAF. In the absence of retinoic acid, a corepressor complex is recruited, composed of N-CoR (nuclear receptor corepressor) or SMRT (silencing mediator of retinoid and thyroid receptors), which binds to another protein called Sin3 and then to HDAC1. HDAC1 is a histone deacetylase protein, which epigenetically alters histones to keep DNA in an untranscribable form. The expression of retinoic acid–responsive genes is essential for normal myeloid development.
Much evidence supports the hypothesis that the PML-RARA fusion protein functions as a dominant negative inhibitor of the PML protein and RXR. PML proteins are normally located in macromolecular nuclear organelles, called PML oncogenic domains (PODs). The PML-RARA fusion protein disrupts the PODs, causing normal PML, RXR, and other nuclear proteins to disperse in an abnormal pattern. ATRA and arsenic trioxide (As 2 O 3 ) have shown activity in APL blast cells and in patients with APL, and studies of these drugs have afforded important insights into the mechanisms of leukemogenesis in APL. ATRA and As 2 O 3 appear to act through different biochemical pathways, and promyelocytes resistant to ATRA are often sensitive to treatment with As 2 O 3 . Both drugs induce degradation of the PML-RARA fusion protein but through different mechanisms. Retinoic acid binding to wild-type retinoic acid receptors (RARs) in the cell nucleus causes degradation of the PML-RARA protein through the ubiquitin-proteosome and caspase systems, thus allowing for the terminal differentiation of leukemic promyelocytes. This binding also results in derepression whereby N-CoR is dissociated from the PML-RARA fusion protein and, subsequently, the nuclear coactivator complex is recruited to reverse histone deacetylase-mediated repression. PODs are relocated back to the normal nuclear pattern. By contrast, As 2 O 3 appears to target the PML portion of the PML-RARA fusion protein preferentially and causes its degradation by inducing apoptosis, potentially through downregulation of the antiapoptotic factor BCL2. The activity of these agents has led to the inferences that fusion proteins may interfere with normal myeloid cell development in several ways—through inhibitory effects on assembly of the PODs that contain PML or through dominant inhibitory effects on transcriptional targets of dimeric complexes with normal retinoid receptors, leading to arrest of differentiation at the promyelocyte stage.
Studies using transgenic mice have shown that the PML-RARA fusion product is involved in the development of leukemia. Transgenic mice generated with the PML-RARA fusion protein specifically expressed in the myeloid-promyelocytic lineage develop a myelodysplastic-like disorder during the first year of life. A subset of these mice then develops a form of acute leukemia that closely mimics human APL and responds to ATRA. Given the relatively long latency period and the development of leukemia in only a fraction of the mice, it is likely that the PML-RARA translocation is insufficient by itself to cause APL and that second mutations are necessary for leukemic transformation.
Mixed-Lineage Leukemia Gene Translocations.
Transcriptional coactivators and corepressors have been implicated in leukemogenesis. An example is the myeloid-lymphoid or MLL protein, a large protein containing transcriptional activation and repression domains, which is believed to act epigenetically to modulate gene expression through its effects on chromatin structure and configuration. Wild-type MLL is required for normal hematopoiesis and HSC development. Mice deficient in MLL die on embryonic day 10.5 and have numerous skeletal, neural, and hematologic deficits. Heterozygous mice have defects in embryonic segmentation and yolk sac hematopoiesis, and adults demonstrate a mild anemia and thrombocytopenia. Chromosomal translocations of the MLL gene on chromosome segment 11q23 have been identified in human AML and ALL and confer a poor prognosis. Many gene partners have been identified that are involved in these translocations, including AF4, AF9, ENL, AF6, ELL , and AF10 . In most cases, the fusion proteins incorporate the 5′ end of MLL and the 3′ end of the partner. Despite the number of different fusion partners, leukemias with MLL translocations tend to possess a distinct genetic signature compared with AML and ALL samples lacking an MLL translocation. This genetic signature includes the upregulation of a number of specific HOX family genes, including HOXA9, HOXA7, and MEIS1 . Wild-type MLL is known to play a major role in HOX gene transcriptional regulation. In turn, HOX genes play critical roles in embryonic development and normal hematopoiesis. A number of studies have clearly demonstrated that overexpression of specific major HOX genes in mouse models and human bone marrow samples, whether by involvement in reciprocal translocations or by gene upregulation, is linked to the transformation of malignant HSCs in AML. The mechanism whereby MLL fusion proteins upregulate HOX gene expression may differ, depending on whether the fusion partner encodes a nuclear partner with direct transcriptional activity or a cytoplasmic protein that results in MLL protein dimerization and subsequent transcriptional activation.
Transcriptional control may be mediated through the modification of histone proteins, which function to maintain chromatin structure. Recently, the DOTL1 complex was identified as a critical factor in MLL-induced leukemogenesis. DOT1L is a histone-3-lysine-79 (H3K79) methyltransferase, which was previously believed to play a fairly ubiquitous role in methylating gene targets in concert with transcription. However, Zhang and Armstrong’s group definitively demonstrated through a series of elegant in vitro and in vivo experiments a specificity for DOT1L in the epigenetic regulation of MLL-translocated downstream targets. When DOT1L was knocked out or absent from MLL-AF9 –transformed cell lines or transgenic mice, leukemia was prevented. The MLL gene is rearranged in up to 20% of pediatric cases of AML. Translocations of MLL are the single most common genetic alteration in infants with acute leukemia, regardless of phenotype, and account for approximately 70% of all cases of AML and ALL in infants. In pediatric and adult cases of AML, the presence of the 11q23 translocation is generally associated with an unfavorable prognosis.
Although less common than MLL translocations, the MLL gene can be altered by partial tandem duplications (PTDs) of particular exons. These PTDs occur in adult and pediatric AMLs with a normal karyotype, as well as those with trisomy 11. Similar to MLL fusions, the MLL -PTD tends to be associated with a poor prognosis and often with the presence of an FLT3 mutation. The mechanisms underlying MLL-PTD leukemogenesis have not yet been elucidated but appear to be different than those of MLL fusions. Interestingly, MLL -PTD appears to repress wild-type MLL expression, which may be an important feature underlying the leukemogenic potential of this abnormality.
Type 2 Mutations.
The leukemogenic mutations discussed in the previous sections involve balanced translocations, duplications, or substitutions that appear to disrupt regulatory mechanisms controlling hematopoiesis and result in dysregulated cell differentiation. Another group of mutations involve unbalanced chromosome rearrangements, such as chromosome loss (e.g., monosomy 5, monosomy 7) or large chromosomal deletions (e.g., 5q−, 7q−, 20q−). These mutations may lead to leukemic transformation by the loss of a tumor suppressor gene (or genes), which then confers a differentiation block and growth advantage to the mutated cells. Identification of these putative tumor suppressor genes has been problematic because the deleted regions are usually large and include many candidate genes. Moreover, leukemogenesis is triggered by the gene’s inactivation rather than by altered structure, making the abnormal gene difficult to identify. However, α-catenin has been identified by our group as a novel tumor suppressor gene on the proximal region of the long arm of chromosome 5. Non–therapy-related AML with these chromosomal losses or deletions occurs most commonly in older patients and is often associated with the prodrome of MDS. In the pediatric population, this class of cytogenetic abnormality is uncommon and is also frequently preceded by MDS. Monosomy 7 is the most frequent chromosomal abnormality documented in children with MDS and is sometimes the sole detectable cytogenetic abnormality. Overall, chromosome loss represents about half of the chromosomal abnormalities identified in MDS.
Therapy-Related AML.
Chromosomal loss and large chromosomal deletions are common in therapy-related AML in the adult and pediatric populations. Although most cases of MDS and AML arise de novo, without evidence of any leukemogenic exposure, in 10% to 20% of patients the disease arises after previous exposure, particularly to topoisomerase II inhibitors, alkylating agents, and ionizing radiation. The 11q23 translocations producing MLL gene fusions are common cytogenetic abnormalities in patients who develop AML after therapy with topoisomerase II inhibitors (e.g., epipodophyllotoxins). AML developing after the use of topoisomerase II inhibitors has a short median latency period of 30 to 34 months, is usually FAB classification M4 or M5, and lacks the antecedent prodrome of MDS. The blast cells of patients with AML or MDS occurring after exposure to alkylating agents often have large chromosomal losses or deletions and have a poor prognosis. The most common chromosomal abnormalities in such cases involve chromosome 7 and chromosome 5. In contrast to the therapy-related AML with MLL fusion genes that develops after treatment with topoisomerase II inhibitors, MDS or AML related to therapy with alkylating agents typically develops after a longer median latency period, 3 to 5 years from the alkylator exposure. Mutations in the TP53 tumor suppressor gene have been previously linked to therapy-related AML and were believed to be infrequent in de novo cases. However, more recently, TP53 mutations have been found to be common in de novo AML with a complex karyotype. In both contexts there is an association with chromosome 5 and 7 copy number alterations.
AML with a Normal Karyotype.
Chromosomal abnormalities have provided some understanding of the mechanisms of malignant transformation in leukemia. However, in many cases of de novo leukemia, no chromosomal abnormality can be detected and the mechanism behind leukemogenesis in these cases is unknown. Because AML involves a block in myeloid differentiation, evidence for mutations in key transcription factors involved in normal myelopoiesis has been sought. Mutations in the early myeloid transcription factors PU.1 and CCAAT/enhancer-binding protein α (C/EBPα) have been identified in a number of AML subtypes and are generally not associated with a known chromosomal translocation. Mouse models have demonstrated that the degree of transcription factor knockdown is critical to the particular phenotype. Mice expressing PU.1 at 20% of normal levels develop AML, whereas those with complete knockdown or 50% of normal levels do not. GATA1, a critical transcription factor for erythrocyte and megakaryocyte development, is mutated in nearly all cases of acute megakaryoblastic leukemia associated with Down syndrome (see later).
Expression profiling using microarrays first used to distinguish prognostic tumor subclasses in breast carcinoma and large B-cell lymphoma has also been applied to leukemia. Using this technology, a set of 50 genes was sufficient to discriminate ALL from AML. More recent validation of this technique in AML has demonstrated the correlation between gene expression profiling studies and the prognostic classification of human AML based on cytogenetic abnormalities. Gene expression profiling has provided valuable new insights into the identification of prognostic subclasses in adult and pediatric AML with a normal karyotype.
Our understanding of the molecular mechanisms underlying de novo AML with a normal karyotype was advanced by the finding of mutations involving the NPM gene in 35% of cases of adult AML and 60% of cases with a normal karyotype. This frequency supersedes the incidence of FLT3 mutations and has established NPM mutations as the most frequent genetic abnormality in adult AML. The frequency of NPM mutations is lower in pediatric AML, occurring in only 6% to 8% of all pediatric patients with AML, but also occurs in a higher number of pediatric patients with a normal karyotype (approximately 27%). The NPM gene on chromosome 5q was previously well known as a partner in a number of oncogenic translocations, including NPM-ALK (anaplastic lymphoma kinase) in anaplastic large cell lymphoma and NPM-RARA in APL. NPM is a molecular chaperone that shuttles between the cytoplasm and nucleus but is found most prominently in nucleoli. It has been shown to function in the prevention of protein aggregation in the nucleolus and the regulation of preribosomal particles through the nuclear membrane. It has also been shown to play a role in the regulation of the alternate reading frame (ARF)-TP53 tumor suppressor pathway. NPM mutations are heterogeneous but occur predominantly in exon 12, uniformly affecting the C-terminal of the protein and causing the formation of a neomorphic nuclear export signal in the resulting NPMc protein. This results in a shift of NPM localization, sequestering it exclusively to the cytoplasm. NPM mutations occur more frequently in children older than 10 years and, as is the case in adults, tend to be associated with a good prognosis. However, there has been a strong association established between patients with NPM mutations and FLT3 -ITDs, which portend a poor prognosis. Patients with both these abnormalities do not fare as well as those with NPM mutations alone.
Similarly, mutations in CEBPA have been associated with a good outcome. CEBPA encodes the CCAAT/enhancer binding protein-alpha (C/EBPα), which functions as a transcription factor that plays a major role in terminal granulocytic differentiation as well as influencing proliferation. C/EBPα is a basic region leucine zipper transcription factor consisting of two amino-terminal transactivation domains, a basic DNA-binding region, and a carboxyl-terminal leucine zipper. Mutations in pediatric AML commonly occur in the amino-terminal domain and bZip domain of the leucine zipper, and many patients harbor both types of mutations.
Mutations in the Wilms tumor 1 (WT1) transcription factor have been identified in approximately 10% of adult and childhood AML, predominantly in patients with a normal karyotype. Mutations are common in exons 7, 8, and 9, comprising three of the four zinc fingers of WT1 . WT1 mutations frequently occur together with FLT3 -ITDs; however, in contrast to the adult studies, they do not independently confer a poor prognosis. Recently, a number of large genomic studies using next-generation sequencing technology revealed novel frequent mutations in DNA methyltransferase 3A (DNMT3A) and IDH1 and IDH2 in adult normal karyotype AML. In adult disease, these mutations confer a poor outcome. By contrast, these mutations are relatively rare in pediatric AML but similarly occur more commonly in patients with a normal karyotype, highlighting the different molecular spectrum of disease in adult and childhood counterparts.
Morphology and Cytochemistry
The initial diagnosis of AML relies on accurate interpretation of the cellular morphology of the bone marrow smears. The diagnosis usually can be made based on the morphologic characteristics of the blasts in a Wright-Giemsa–stained bone marrow smear ( Fig. 51-5 ). In addition to general morphology, the diagnosis can be confirmed in cases exhibiting Auer rods (thin, needle-shaped cytoplasmic deposits that stain pink with Wright-Giemsa and are strongly myeloperoxidase [MPO]-positive). In addition to examination of the morphology of the blasts, the Wright-Giemsa stain is also used to assess for dysplastic features in erythroid, myeloid, and megakaryocytic cell lines.
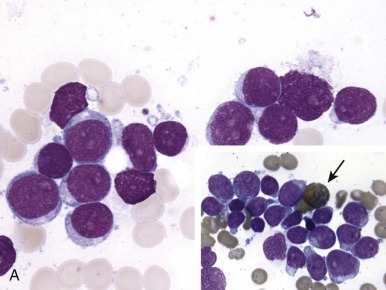
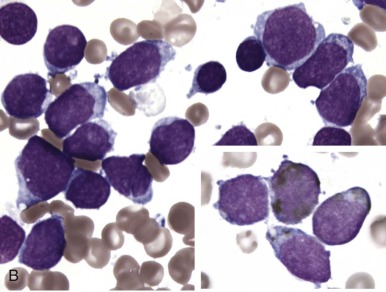
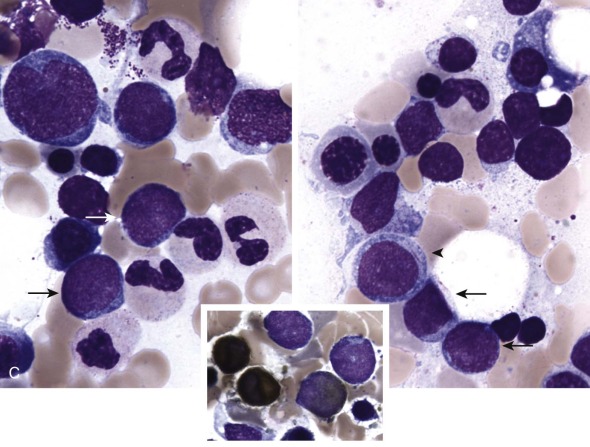
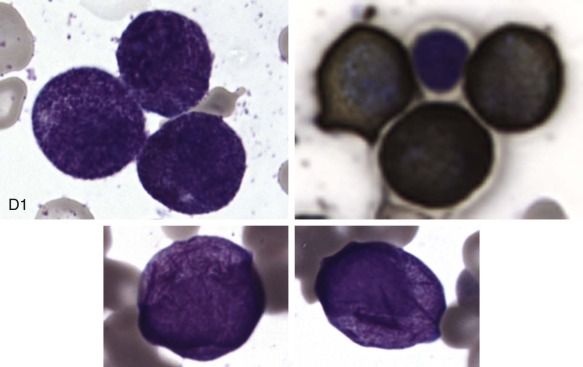
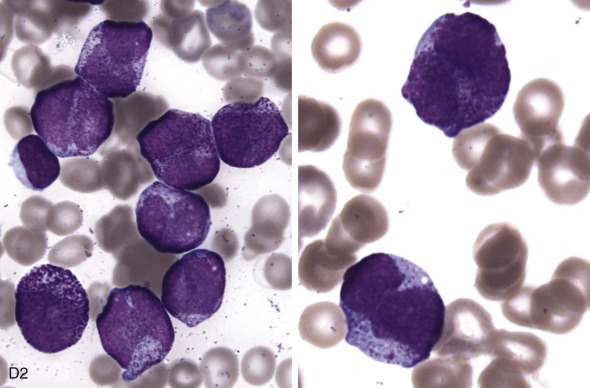
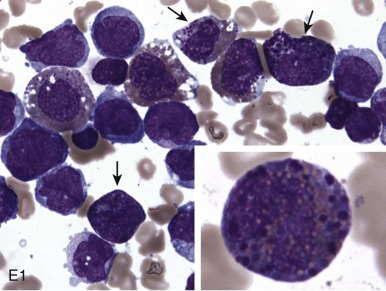
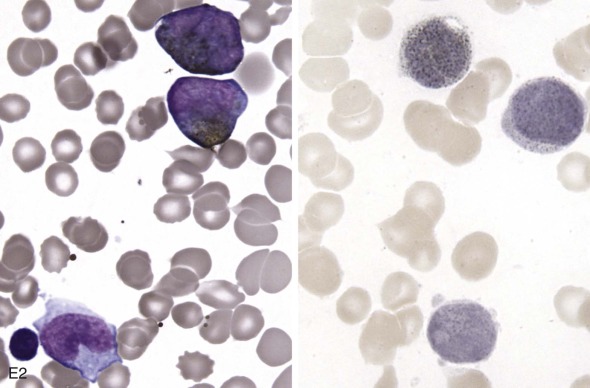
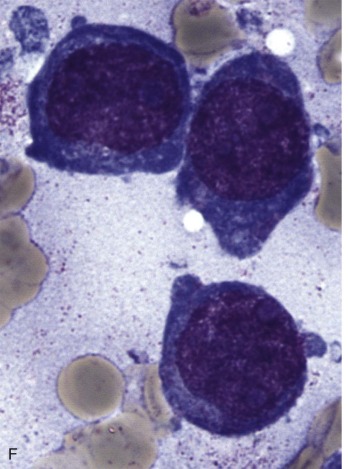
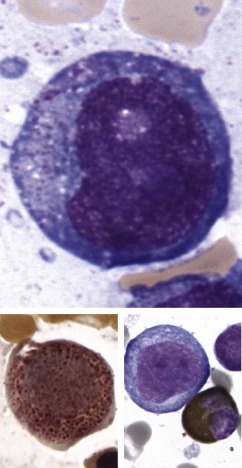
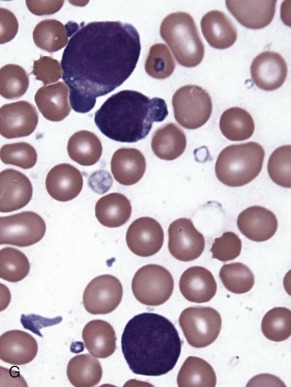
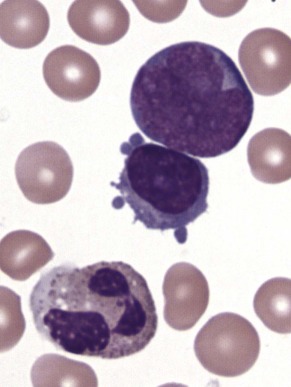
Additional cytochemical stains are important for the accurate diagnosis of AML. MPO and Sudan black B (SBB) are cytochemical stains for granulocyte, eosinophil, and monocyte lineages. Peroxidase is present in the primary granules of myeloid cells, beginning at the promyelocyte stage and throughout subsequent maturation. Typically, leukemic myeloblasts are also strongly peroxidase-positive. MPO staining of Auer rods is particularly robust in leukemic blasts and can demonstrate Auer rods not recognized using the Wright-Giemsa stain. By contrast, the MPO staining of monoblasts shows a fine granular pattern in the monoblast cytoplasm. The staining pattern of SBB is similar to that of peroxidase, particularly in myeloblasts and in the detection of Auer rods. Monoblasts either do not stain or show a weakly positive, diffuse pattern with SBB.
The esterase stains are useful for distinguishing granulocytic cells from cells of the monocytic lineage. Chloroacetate esterase is a specific stain for granulocytes and mast cells. Results of staining of early myeloblasts are often negative, but promyeloblasts and later granulocytic lineage cells stain strongly, as do Auer rods. Monoblasts do not stain with chloroacetate esterase. In contrast, the nonspecific esterases (NSEs) such as α-naphthyl butyrate esterase and α-naphthyl acetate esterase stain monoblasts and monocytes strongly. Granulocytic and lymphoid cells do not stain with the nonspecific esterases. The blast cells of AML-M4 (i.e., myelomonocytic leukemia) should demonstrate positive chloroacetate esterase activity and α-naphthyl butyrate esterase or α-naphthyl acetate activity; this finding is important for making the diagnosis. The periodic acid–Schiff (PAS) reaction is not as informative as the previously described stains, but it can be useful, particularly in the diagnosis of AML-M6 (i.e., erythroleukemia). Leukemic erythroblasts can stain strongly with PAS, whereas normal erythroid precursors generally demonstrate no PAS activity.
Immunophenotype
Since the 1980s, multiparameter flow cytometric analysis has become an important element in the diagnosis and classification of leukemia. More recently, multiparameter flow cytometry has been used as a tool to detect MRD in acute myeloid leukemia and is being incorporated into clinical trials to clarify prognostic groups and help make treatment decisions. Immunophenotyping using a large panel of monoclonal antibodies to myeloid and lymphoid lineage and progenitor cell–associated antigens has been used to discriminate myeloid or lymphoid differentiation correctly in up to 98% of patients. Monoclonal antibodies have been classified based on their reactivity with the lineage or differentiation-associated antigens on the surfaces of normal and malignant cells. Because the monoclonal antibodies are not leukemia cell–specific and most of the hematopoietic antibodies are not strictly lineage specific, it is necessary to use panels of antibodies on blast cell populations and to incorporate the immunophenotypic information with the clinical findings and other morphologic, cytochemical, and cytogenetic results for accurate leukemia cell classification.
Blasts of myeloid origin generally express HLA-DR, CD33, and CD34. Monocytic cells usually express HLA-DR at all stages of maturation, whereas promyelocytes and more mature cells of the granulocyte lineage do not. CD33 and CD13 are expressed on neutrophil and monocyte precursors, but CD33 is absent on mature neutrophils. CD14 is relatively specific for cells of the monocytic lineage. As myeloid differentiation continues, CD15 expression increases, whereas CD34 expression decreases. Megakaryocytic cells and platelets express CD41, CD42, and CD61.
Immunophenotyping has been particularly valuable for lineage assignment in undifferentiated leukemias (i.e., AML-M0), identification of acute megakaryocytic leukemia, and analysis of the lineage of leukemias with MLL gene translocations.
Cytogenetics
Cytogenetic analysis is an essential component in the diagnosis and treatment of AML. The chromosomal aberrations detected by cytogenetic testing also have prognostic value and may be used as tumor markers. In the WHO classification of hematologic malignancies, specific cytogenetic abnormalities have been included in the diagnostic and prognostic criteria for differentiating subclasses of AML.
Cytogenetic analyses of bone marrow samples have revealed chromosomal abnormalities in most patients with AML. In the pediatric population, the incidence of cytogenetic abnormalities is even higher than that in adults. However, the distribution of the specific cytogenetic abnormalities is different in the various age groups. In infants with AML, the most common chromosomal aberrations are translocations involving the MLL gene (band 11q23). The incidence of 11q23 translocations is as high as 40% in AML blasts of children younger than 2 years. After age 2 years, the frequency of 11q23 translocations decreases with age, and the translocation is detectable in AML blasts from less than 10% of older children and adults. One rare chromosomal abnormality, the t(1;22)(p13;q13), is found almost exclusively in non–Down syndrome infants with acute megakaryocytic leukemia. In a published review of 39 patients with this translocation, 95% were younger than 2 years, and it was not identified in adults with AML. This translocation is now known to result in aberrant expression of the OTT-MAL fusion gene.
There are two classification systems for AML (see next section). The FAB classification is based exclusively on morphology. However, there are some morphologic subtypes such as M3 and M4eo that are always associated with particular cytogenetic changes, that is, RARA rearrangements for the former and inv(16) or t(16;16) in the latter. In addition, there are certain cytogenetic changes that are often (if not exclusively) associated with a particular subtype and that carry prognostic significance, such as the good prognosis t(8;21) often associated with the M2 subtype or MLL rearrangements often associated with the M5 subtype. The WHO classification system takes this into account and has a category of “acute myeloid leukemia with recurrent genetic abnormalities,” which includes t(8;21), inv(16) or t(16;16), t(15;17), and 11q23. The fusion proteins thus generated are discussed in more detail earlier (see “Acute Myelogenous Leukemia: Biology”). Some of the more common chromosomal abnormalities and their associated FAB and WHO subtypes are shown in Table 51-1 and Box 51-2 . The prognostic significance of certain cytogenetic abnormalities is discussed later (see “ Prognosis ”).
| FAB Subtype | MPO | SBB | NSE | PAS | MYELOID | ERYTHROID | MEGAKARYOCYTIC | T-CELL LINEAGE | OTHER | Translocations and Rearrangements | Genes Involved | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD 11b | CD 13 | CD 14 | CD 15 | CD 33 | CD 34 | CD 65 | Glycophorin A | CD 41/61 | CD 42 | CD 36 | CD 2 | CD 3 | CD 4 | CD 7 | CD 56 | HLA-DR | CD 117 | |||||||
| M0 | − | − | − | − | − | ++ | + | + | ++ | ++ | + | + | + | + | − | + | − | + − | + − | + | ++ | ++ | 11q23 Trisomy 4, 8,13, −5, −7 | MLL rearrangements |
| M1 | + | + | − | − | − | + | − | + | + | + | − | − | − | + | − | + | − | − | + | + | ++ | ++ | t(8;21)(q22;q22) t(9;22)(q34;q11) Trisomy 8, −5, −7 | AMLI-ETO BCR-ABL |
| M2 | + | + | − | − | + | + | − | + | ++ | ++ | − | − | − | + | − | + | − | − | + | + | ++ | + | t(8;21)(q22;q22) t(6;9) inv3(q21;q26), t(3:3) (q21;q26) Trisomy 8, −5, −7 | AMLI-ETO DEK-CAN EVI1 |
| M3 | + | + | − | − | + | + | − | + | + | + | − | − | − | − | + | − | + | + | + | + | t(15;17)(q22;q12) t(11;17)(q23;q12), t(5;17) (q32;q12) | PML-RARA PLZF-RARA NPM-RARA | ||
| M4 | + | + | + | − | ++ | ++ | ++ | + | + | + | − | − | − | ++ | − | + | − | ++ | + | + | +++ | + | t(6;9) inv3(q21;q26), t(3:3) (q21;q26) t(9;11)(p22;q23) 11q23 | DEK-CAN EVII AF9-MLL MLL rearrangements |
| (M4eo) | + | + | + | + | ++ | + | + | + | + | + | − | − | − | ++ | − | − | +++ | + | inv16(p13;q22), t(16:16) | MYHII-CBFβ | ||||
| M5 | − | − | + | − | ++ | ++ | ++ | ++ | + | ++ | − | − | − | ++ | − | + | − | ++ | + | + | +++ | + | t(11;17)(q23;q21) 11q23 11q23,t(9;11)(p22;q23) [M5a] t(8;16), t(10;11)(p12;q23) [M5b] | MLL-AF17 MLL rearrangements MLL, AF9-MLL MOZ-CBP, AF10-MLL |
| M6 | + | + | − | + | − | + | − | − | + | + | ++ | − | − | − | − | − | + | + | − | + | Trisomy 8, −5, −7 | |||
| M7 | − | − | + | + | − | + | − | − | + | + | − | ++ | ++ | + + | ++ | − | + | + | + | + | + | t(1;22)(p13;q13) | OTT-MAL | |
| M7 (DS) | − | − | + | + | − | + | − | − | + | + | − | ++ | ++ | ++ | ++ | − | + | + | + | + | + | Trisomy 21 | GATA-1 | |
* Shown in boldface.
Acute Myeloid Leukemia with Recurrent Genetic Abnormalities (FAB)
t(8;21)(q22;q22); (AML1/ETO; RUNX1-RUNX1T1) (M2)
inv(16)(p13;q22) or t(16;16)(p13;q22); (CBFβ/MYH11) (M4E0)
t(15;17)(q22;q12) (PML/RARA) (M3)
AML with t(9;11)(p22;q23); MLLT3-MLL
AML with t(6;9)(p23;q34); DEK-NUP214
AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1
Acute Myeloid Leukemia with Myelodysplasia-Related Changes
Following myelodysplastic syndrome (MDS) or MDS/myeloproliferative disease (MPD)
Therapy-Related Myeloid Neoplasms
Acute myeloid leukemia (FAB)
Acute myeloid leukemia, minimally differentiated (M0)
Acute myeloid leukemia without maturation (M1)
Acute myeloid leukemia with maturation (M2)
Acute myelomonocytic leukemia (M4 )
Acute monoblastic and monocytic leukemia (M5)
Acute erythroid leukemia (M6)
Acute megakaryoblastic leukemia (M7)
Acute basophilic leukemia
Acute panmyelosis with myelofibrosis
Myeloid Sarcoma
Myeloid Proliferations Related To Down Syndrome
Blastic Plasmacytoid Dendritic Cell Neoplasm
French-American-British Classification
In 1976, the FAB group proposed a system of classification of AML based on morphologic and cytochemical features (see Table 51-1 ). The system divides AML into seven subtypes, M1 through M7. An M0 subtype was later added to describe undifferentiated leukemia and, since 1976, immunophenotypic data have also been included. In general, AML can be differentiated from ALL based on morphologic features and cytochemical stains. Cells of myeloid origin should stain with myeloperoxidase and SBB. Cells of monocytic derivation usually stain with NSE. AML blasts usually do not have PAS activity, except for erythroblasts in AML-M6 and eosinophils of the M4Eo subtype.
M0: Acute Myeloblastic Leukemia with Minimal Differentiation.
In patients with AML-M0, the bone marrow is usually hypercellular and more than 90% of the cells are blasts. Most blast cells lack cytoplasmic granules, nucleoli, or Auer rods, and results with MPO and SBB stains are negative. Immunophenotypic analysis shows the presence of myeloid or monocytic cell antigens (i.e., CD13, CD14, CD33, or CD34) that are detectable on the cell surface of most AML-M0 blasts, but some AML-M0 blasts express terminal deoxynucleotidyl transferase, an enzyme usually associated with ALL. AML-M0 is associated with a high incidence of cytogenetic abnormalities, most of which are complex and often involve chromosomes 5 and 7, trisomy 8, or MLL rearrangements.
M1: Acute Myeloblastic Leukemia without Maturation.
The bone marrow of patients with AML-M1 is hypercellular and filled with myeloblasts (more than 90% blasts). Most blasts stain with MPO and SBB. The blast cells in this subtype display minimal myeloid differentiation. Morphologically, the blast cells may contain scant gray-blue cytoplasm and few or no azurophilic granules or Auer rods. Prominent nucleoli are usually detectable. Flow cytometric analysis usually shows that the blast cells of AML-M1 express HLA-DR, CD13, CD33, and CD34. Cytogenetic abnormalities for this subclass often include monosomy 5 or 7 or trisomy 8.
M2: Acute Myeloblastic Leukemia with Maturation.
The bone marrow of patients with AML-M2 usually shows evidence of some maturation beyond the myeloblast, with evidence of maturation beyond the promyelocyte stage in more than 10% of nonerythroid cells. Myeloblasts must represent more than 30% of the bone marrow cells but less than 90% of nonerythroid cells. The blasts generally have a few clusters of primary granules and stain with MPO and SBB. Auer rods and prominent nucleoli are common. Immunophenotypic expression of HLA-DR, CD13, CD33, CD11, and CD15 is typical. This subtype may exhibit monosomy 5 or 7, trisomy 8, t(8;21), t(6;9), or abnormalities of chromosome 3.
M3: Acute Promyelocytic Leukemia.
There are two types of AML-M3. The more common type is the hypergranular variant, in which more than 30% of the blasts are promyelocytes and myeloblasts. Most promyelocytes have heavy granulation. Auer rods and Auer rod bundles are common. In the rare microgranular variant, the cells exhibit fine cytoplasmic granules, which are not distinguishable by light microscopy and nuclear morphologic irregularities (i.e., microgranular M3 or M3v). The cells of both subtypes stain strongly with MPO and SBB and also with chloroacetate esterase. The promyelocytes usually do not show PAS and NSE activity. The blasts express CD13, CD33, CD11, and CD15, but they are HLA-DR− and CD14−. This subtype exclusively exhibits RARA rearrangements, the vast majority of which are t(15;17) leading to PML-RARA .
M4: Acute Myelomonocytic Leukemia.
The M4 subtype is defined by the presence of blast cells with both granulocytic and monocytic features. The bone marrow in these patients has more than 30% infiltration by immature myeloid precursors, and there is often extramedullary involvement and a peripheral blood monocytosis. The blasts are usually pleomorphic with regard to size, amount of cytoplasm, granularity, and nuclear morphologic features. Some Auer rods can be seen, and prominent nucleoli are usually present. Staining is variable, with some of the blast cells showing positivity for MPO, SBB, and NSE. M4 AML may be associated with t(6;9), abnormalities of chromosome 3, and MLL gene rearrangements, in particular t(9;11). A small proportion of patients with AML-M4 have a moderate eosinophilia in their bone marrow (M4Eo). The eosinophils are notable for the presence of basophilic and eosinophilic granules and stain with chloroacetate esterase and PAS. Cell surface antigen expression includes CD13, CD15, CD33, CD4, CD11c, CD14, CD64, and HLA-DR. M4eo AML exhibits an inv(16) or t(16;16).
M5: Acute Monocytic Leukemia.
More than 80% of the nonerythroid bone marrow cells in patients with AML-M5 are monocytic. There are two subtypes: M5a (undifferentiated) and M5b (differentiated). In AML-M5a, more than 80% of the cells are monoblasts. Patients with AML-M5a tend to be younger, have higher presenting white blood cell (WBC) counts, and have a poorer prognosis. In AML-M5b, fewer than 80% of the monocytic cells are monoblasts, and most of the cells are recognizable as monocytes or promonocytes. The peripheral blood in patients with AML-M5b exhibits a profound monocytosis. In both subtypes, the blasts demonstrate strong NSE activity. MPO and SBB staining results are usually negative. Cell surface antigen expression includes CD13, CD15, CD33, CD4, CD11c, CD14, CD64, and HLA-DR. M5 AML often exhibits MLL gene rearrangements, t(9;11) or t(11;17) in M5a and t(10;11) in M5b.
M6: Acute Erythrocytic Leukemia.
AML-M6 is an uncommon form of AML overall and is rare in children. This form of leukemia is defined by a more than 50% erythroblast infiltration of the bone marrow. M6 AML may exhibit monosomy 5 or 7 or trisomy 8.
M7: Acute Megakaryoblastic Leukemia.
The bone marrow exhibits an infiltration with pleomorphic megakaryoblasts that often display cytoplasmic budding and may appear in clusters. Aspiration of the bone marrow may be challenging due to associated fibrosis. M7 blasts generally do not stain with MPO and SBB but show activity with PAS. Immunophenotyping is usually required to distinguish AML-M7 from ALL-L2, documenting the presence of the megakaryocytic antigens, glycoprotein Ib, glycoprotein IIb/IIIa, or factor VIII. Non–Down syndrome infants with M7 AML often exhibit a t(1;22). Down syndrome patients have a very high incidence of M7 AML with a particularly good prognosis, which is in contrast to the poor prognosis of M7 AML in non–Down syndrome children and adults.
World Health Organization Classification
The discovery of cytogenetic and molecular genetic abnormalities in malignant disease has had a significant impact on our understanding of malignant transformation. In myeloid malignancies, some genetic abnormalities have prognostic significance whereas others appear to define specific disease subtypes. The WHO has proposed a classification system for neoplastic diseases of the hematopoietic and lymphoid tissues that includes a classification for AML (see Box 51-2 and Fig. 51-5 ). This classification uses the traditional FAB-type morphologic categories of disease and includes additional entities such as immunophenotype, molecular genetic, and clinical characteristics that make the system more clinically relevant for diagnosis, prognosis, and treatment. The WHO classification system divides myeloid diseases into several major subtypes—myeloproliferative neoplasms; myeloid neoplasm associated with eosinophilia and abnormalities of PDGFRA , PDGFRB , or FGFR1 ; myelodysplastic-myeloproliferative neoplasms; MDS, AML and related neoplasms; and acute leukemias of ambiguous lineage. The 2008 revision of the WHO classification has expanded the category of AML to include AML with recurrent cytogenetic translocations, AML with myelodysplasia-related changes, therapy-related myeloid neoplasms, AML not otherwise specified, myeloid sarcoma, myeloid proliferations related to Down syndrome, and blastic plasmacytoid dendritic cell neoplasms.
Within the subtype containing recurrent cytogenetic translocations, seven additional categories have been described:
AML with t(8;21)(q22;q22); AML1-ETO ; RUNX1-RUNX1T1
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11
APL with t(15;17)(q22;q12); PML-RARA
AML with t(9;11)(p22;q23); MLLT3-MLL
AML with t(6;9)(p23;q34); DEK-NUP214
AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1
AML with t(6;9)(p23;q34) and AML with inv(3)(q21q26.2) is relatively uncommon in children. Myeloid sarcomas, formerly called chloromas, may precede hematopoietic evidence of disease and so are now classified as a separate entity. The myeloid diseases of Down syndrome were incorporated into the revised WHO classification in 2008 and are described later in this chapter. Plasmacytoid dendritic cell neoplasms were formerly referred to as blastic NK-cell lymphomas, but the nomenclature has changed to reflect the origin of this myeloid malignancy in this subset of dendritic cells. This rare disease can manifest in children with skin lesions, adenopathy, and ultimately bone marrow involvement. Blast cells characteristically express CD4, CD43, CD56, and CD123 without expression of CD34 or CD117.
The FAB standard used to define AML had been 30% replacement of the bone marrow by the blast cells, but later studies have shown that patients with 20% to 30% blasts (previously classified as refractory anemia with excess blasts in transformation [RAEB-T]) have a prognosis similar to that of patients with more than 30% blasts. The blast count for the diagnosis of AML in the WHO classification was therefore changed to 20%, and the category of RAEB-T was eliminated.
Certain specific and recurrent cytogenetic abnormalities are common in MDS, in alkylating agent–related AML, and in de novo AML with a poor prognosis. These abnormalities include 3q−, −5, 5q−, −7, 7q−, +8, +9, 11q−, 12p−, −8, −19, 20q−, +21, t(1;7), t(2;11), and complex karyotypes. In the classification scheme, these cytogenetic abnormalities were considered to indicate a poor prognosis. Prior therapy with topoisomerase II inhibitors (i.e., epipodophyllotoxins and doxorubicin) was also associated with a poor prognosis. Typically, patients in whom AML develops after exposure to these agents have translocations involving 11q23 ( MLL ). The WHO classification includes these patients in the poor-prognosis category but distinguishes them from having alkylating agent–related secondary leukemia.
Patients may have MDS with unilineage or multilineage dysplasia, and these are reflected as distinct diseases in the most recent iteration of the WHO classification. Dysplasia is defined as being observed in greater than or equal to 10% of the cells of a given myeloid lineage. Children with 2% to 19% peripheral blasts or 5% to 19% bone marrow blasts can be classified according to the same criteria as adult MDS, but a new category of refractory cytopenia of childhood has been introduced to define children with persistent cytopenias and dysplasia of two or more lineages but less than 2% peripheral blasts or less than 5% bone marrow blasts.
Clinical Presentation
The presenting signs and symptoms of AML result from leukemic blast cell infiltration of the bone marrow. The leukemia cells overwhelm the processes of normal hematopoiesis, resulting in anemia, thrombocytopenia, and neutropenia. It is rare for AML to be diagnosed incidentally on a routine medical evaluation, but there is considerable variability in the range of presenting signs and symptoms in patients with de novo AML. Usually, patients seek medical attention for fever, fatigue, pallor, skin or mucosal bleeding, bone pain, or infections not responding to appropriate antibiotic therapy. Bone pain is a common symptom, and patients may present with a limp, rib pain, or back pain.
The WBC count of patients with newly diagnosed AML can range from less than 1,000/µL to more than 500,000/µL. The leukocyte count is more than 100,000/µL in approximately 25% of pediatric patients with AML, and an elevated count is more common in those with the M4 and M5 subtypes and in those with FLT3 -ITD mutations. The number of circulating granulocytes is often critically decreased, regardless of the total leukocyte count, and, because their function is usually impaired, the risk for overwhelming bacterial infection in patients with newly diagnosed AML is markedly increased. Patients with AML who have a fever require immediate treatment with broad-spectrum antibiotics after appropriate culture samples have been obtained. The hemoglobin level is occasionally normal but is usually less than 9 g/dL, and levels as low as 3 g/dL at diagnosis are not uncommon. About half of children with new-onset AML have platelet counts of 50,000/µL or less, which increases the risk for life-threatening bleeding.
Disseminated intravascular coagulation (DIC) has been observed in patients with all FAB subtypes of AML but is most common in acute promyelocytic leukemia (APL; see later). Before the development of ATRA, treatment with low-dose heparin during induction was initiated for patients with AML-M3 to prevent DIC. The later addition of ATRA to induction therapy rapidly resolved DIC in patients with AML-M3 and markedly reduced early mortality.
Patients with hyperleukocytosis have an increased risk for mortality from leukostasis, particularly in the brain or lung, and urgent leukapheresis or treatment with leukocyte-reducing agents such as hydroxyurea may be necessary (see “Complications”). Extramedullary leukemia occurs in 20% to 25% of children with AML and may include chloromas (tumor nodules), skin infiltration, CNS disease, gingival infiltration, hepatosplenomegaly, or testicular involvement. Chloromas (e.g., myeloblastomas and granulocytic sarcomas) are solid tumors of myeloblasts that may occur in any area of the body but are most common in the orbit and epidural area. Skin infiltration, or leukemia cutis, usually presents as slightly purple lesions (i.e., “blueberry muffin” spots), is more common in infants with monocytic leukemia, and may be the initial sign of disease. CNS involvement at diagnosis is more common in AML than in ALL. Up to 15% of patients with AML have myeloblasts in the cerebrospinal fluid at diagnosis. Gingival infiltration may present as hyperplasia, often accompanied by bleeding. Severe extramedullary disease with massive hepatosplenomegaly is also a more common finding in infants. Extramedullary involvement of the testes can also be documented but is less common than in ALL. Extramedullary infiltration, particularly gingival involvement, is more common with the M4 and M5 (acute myelomonocytic and acute monoblastic and monocytic) leukemias. Chloromas and CNS disease are more common in AMLs with t(8;21). Extramedullary infiltration in pediatric patients is not clearly associated with worse prognosis unless accompanied by a high presenting WBC count.
Treatment
As is true for any pediatric cancer, the goal of therapy for children with AML is to completely eradicate the disease while limiting treatment-induced toxicity. In general, therapy for AML includes immediate supportive care measures followed by intensive remission induction and consolidation chemotherapy. At any given time, several large, multicenter phase III clinical trials are ongoing, each aimed to improve overall survival (OS) through randomized comparisons of treatment options while minimizing short- and long-term toxicities.
Remission Induction
During the past 40 years, survival rates for patients with AML have risen from less than 10% to more than 50% of patients because of intensification of chemotherapy, use of hematopoietic stem cell transplantation (HSCT), and improved supportive care. The first phase of chemotherapy is termed induction and consists of two to four cycles of intensive chemotherapy with the goal of inducing remission. Morphologic remission is defined as the presence of fewer than 5% blasts visible on bone marrow aspirate obtained after recovery of counts. Patients may also achieve cytogenetic and FISH-negative remission, as defined by the absence of a previously detected leukemic clone–associated cytogenetic abnormality in the bone marrow cells. Remission is also now being defined by the absence of MRD, assessed by multiparameter flow cytometry that is designed to detect as few as 0.1% leukemic blasts in the marrow. The inclusion of MRD evaluation has not only identified children at risk for relapse who appeared to be in morphologic remission but has also revealed false-positive recovering marrows that had been deemed refractory disease.
Induction chemotherapy should be initiated as soon as the diagnosis of AML is confirmed, preferably by morphologic, immunophenotypic, and cytogenetic studies of the bone marrow. In some cases, particularly in patients presenting with hyperleukocytosis, the patient’s condition may be too unstable for bone marrow evaluation, in which case the diagnosis can often be confirmed on studies of the peripheral blood. Induction chemotherapy regimens consist of high-dose myelosuppressive cytotoxic agents that result in prolonged pancytopenia. The period of bone marrow hypoplasia generally lasts from 21 to 30 days from the initiation of therapy but may last considerably longer. During this period, patients have a high risk for life-threatening infection and bleeding. Aggressive supportive care is necessary (see later).
Historical North American Approaches to Treating AML.
The basis of modern induction therapy for AML is a combination of cytarabine and an anthracycline. The classic combination therapy was the so-called 7 + 3 regimen, which consists of a continuous 7-day intravenous infusion of cytarabine at 100 to 200 mg/m 2 /day and 3 days of bolus infusions of daunorubicin at 45 to 60 mg/m 2 /day. Remission is achieved in 60% to 70% of patients with newly diagnosed AML using this regimen. In children, several strategies have been used to intensify this regimen to achieve higher rates of CR. CCG trial 213 added etoposide, thioguanine, and dexamethasone to the traditional 7 + 3 regimen to create the five-drug Denver regimen. The Denver and 7 + 3 regimens were compared in a randomized fashion, and there was no significant difference in CR rate, with 79% of children achieving CR with 7 + 3 and 76% achieving CR with Denver regimen.
In CCG trial 2891, the same drugs were used but the timing was intensified in an attempt to improve CR. The hypothesis was that leukemia cells may be recruited synchronously into the cell cycle after chemotherapy, and therefore reexposure at an earlier time point may affect cells while more of them are at a sensitive phase in the cell cycle. Thus the Denver regimen was modified into the five-drug DCTER regimen, again consisting of d examethasone, c ytarabine, t hioguanine, e toposide, and R ubidomycin (daunorubicin). Induction consisted of a total of four cycles of chemotherapy, and patients were randomized to standard or intensive timing. For standard timing, each cycle of induction began with count recovery from the previous cycle. With intensive timing, cycles 2 and 4 began at day 10 after the start of cycles 1 or 3, respectively, regardless of hematologic status. Not surprisingly, the intensive timing resulted in far greater toxicity, with 11% of patients in this arm of the trial dying of toxicity compared with only 4% of patients dying of toxicity in the standard timing trial arm. Because of this, better supportive care measures were instituted for the intensive timing trial arm, including more aggressive empiric antibiotic and antifungal coverage and the use of granulocyte colony-stimulating factor (G-CSF). Interestingly, the CR rates were similar for the two arms in the trial—75% for intensive timing and 70% for standard timing—but the reason for failure to achieve remission was different, with 11% toxic death and only 14% resistant disease in the intensive timing arm versus only 4% toxic death but 26% resistant disease in the standard timing arm. The most dramatic results, however, came later, when it was shown that for patients who achieved a CR there was a 3-year disease-free survival (DFS) of 55% for those who had been treated in the intensive timing arm, versus a DFS of only 37% for patients who had received standard timing. For this reason, the standard timing arm of the trial was closed early and an intensive timing DCTER/DCTER schedule became the new standard combination therapy for newly diagnosed AML patients.
In the meantime, a comprehensive review of five randomized trials comparing idarubicin with daunorubicin as induction therapy for AML had shown improved remission induction rates and OS for patients receiving idarubicin. For this reason, the CCG pilot trial 2941 tried to intensify therapy further by using idarubicin in place of daunorubicin in each cycle of the intensively timed regimen. Because of unacceptably high toxicity, with a toxic death rate of 14%, this was modified to idarubicin only in cycles 1 and 3, and more strict supportive care guidelines were put into place, including mandatory hospitalization during neutropenia, and stricter infection prophylaxis (see later, “ Supportive Therapy at Diagnosis and during Therapy ”). The addition of idarubicin showed no advantage in remission induction or event-free survival (EFS) but did show a decrease in marrow blasts at day 14 of induction. CCG trial 2961 continued to use the hybrid idaDCTER/DCTER intensive timing induction regimen resulting in induction remission rates of 89%, which were similar to historic controls. Most of the improvement was believed to be related to better supportive care rather than to the change in chemotherapy.
Medical Research Council Trials (MRC10, MRC12, and MRC15).
During this period, data emerged from the British MRC trials that seemed to show better survival and less toxicity. MRC10 induction chemotherapy randomized DAT (daunorubicin, cytarabine, thioguanine) to ADE (cytarabine, daunorubicin, etoposide) in a 10-day cycle of chemotherapy followed by an 8-day cycle of the same drugs on WBC count recovery. The CR rate for children in this study was 93%, with a 4% rate of toxic death and a 3% rate of resistant disease. Early data suggested a benefit for pediatric patients using ADE versus DAT, but the later data showed no difference in 10-year OS (DAT, 57%; ADE, 51%; P = .3), DFS (DAT, 53%; ADE, 48%; P = .3) or EFS (DAT, 48%; ADE, 45%; P = .5). Because of the early results possibly favoring ADE, MRC12 compared ADE with MAE (mitoxantrone, cytarabine, etoposide) in a 10-day cycle followed by an 8-day cycle on count recovery. The CR rates were similar for the two groups (ADE, 92%; MAE, 90%; P = .3) with a slight increase in toxic deaths for the MAE group (ADE, 3%; MAE, 6%) but a lower relapse rate (ADE, 39%; MAE, 32%) and better DFS rate for the MAE group (ADE, 55%; MAE, 63%), although the 10-year EFS and OS rates were not significantly different (54% and 63%, respectively). In MRC15 there were three randomized induction backbones—ADE; cytarabine and daunorubicin without etoposide, or idarubicin, fludarabine, cytarabine, and G-CSF (Ida-FLAG)—and three randomized consolidation backbones—amsacrine, cytarabine, and etoposide; cytarabine 1.5 g/m 2 ; or cytarabine 3 g/m 2 with and without the addition of gemtuzumab ozogamicin (GMTZ, GO, Mylotarg). GMTZ, a recombinant humanized anti-CD33 monoclonal agent linked to the tumor antibiotic calicheamicin, was incorporated as a targeted therapy in these studies. Because CD33 is expressed on most AML cells but not on pluripotent HSCs or other tissues, this drug can target leukemia cells more specifically. GMTZ was well tolerated overall, but no overall improvements in survival or relapse rate were observed regardless of the chemotherapy backbone in either induction or consolidation.
Recent U.S. Trials.
The next series of U.S. trials built on the data from these recent MRC studies. The COG has undertaken three therapeutic trials to date based on an MRC backbone. The COG AAML03P1 pilot and AAML0531 phase III groupwide study backbone included cytarabine/daunorubicin/etoposide (ADE 10+3+5) followed by ADE 8+3+5, cytarabine/etoposide (AE), mitoxantrone/cytarabine (MA), and, finally, high-dose cytarabine (Capizzi II) for children who did not go on to receive HSCT. The COG pilot trial AAML03P1 assessed the safety of adding a single dose of GMTZ to ADE 10+3+5 and MA cycles. Toxicity on this pilot study was acceptable, so GMTZ was added to these same two cycles on AAML0531 in a 1 : 1 randomization. Subsequent outcome data from the AAML03P1 trial demonstrated a 3-year EFS of 53% and OS of 66% with no increased treatment-related mortality over backbone chemotherapy. Outcome data have not yet been released for AAML0531. However, after data were released from the Southwest Oncology Group (SWOG) Study 106, the U.S. Food and Drug Administration (FDA) removed GMTZ from the U.S. market because this adult-based study showed no benefit in outcome to the addition of GMTZ but an increase in induction deaths. By contrast, subgroup analysis on the MRC 15 trial found that the addition of GMTZ to induction chemotherapy was of benefit to CBF AML and to intermediate-risk patients and did not contribute to increased toxicity. Data from the COG AAML0531 trial demonstrated mixed results. GMTZ improved 3 year EFS and relapse risk for all patients but not OS. Interestingly, positive trends were seen for GMTZ in subgroup analysis, including OS in high risk patients and relapse risk in low risk patients, although this was also associated with increased TRM in the low risk subgroup. Given this controversy regarding both toxicity and efficacy and the absence of data from the AAML0531 at the time of study initiation, GMTZ was not included on the current COG AAML1031 trial. However, other co-operative groups are planning future trials to examine the optimal dosing of GMTZ in pediatric AML (A. Gamis, personal communication, July 2014). The current COG trial risk stratifies patients into high- and low-risk AML based on cytogenetics and MRD and randomizes the addition of the proteosome inhibitor bortezomib as a potential AML stem cell targeted therapy. In addition, patients harboring the FLT3 -ITD with sufficient allelic ratio have the option of being nonrandomly assigned to an MRC-based backbone arm with the addition of the FLT3 inhibitor sorafenib. Given recent MRC data that four cycles appear as effective as five cycles of therapy, only four cycles are included on this protocol (ADE 10+3+5; ADE 8+3+5; cytarabine and etoposide; and mitoxantrone and cytarabine) for good responders. Patients with positive MRD after cycle 1, defined as more than 0.1% by multiparameter flow cytometry (MPFC) receive mitoxantrone and cytarabine instead of an additional ADE cycle, and high-risk patients receive high-dose cytarabine for cycle 4.
The St. Jude Children’s Research Hospital (SJCRH) trial AML02 randomized patients to standard low- or high-dose cytarabine during the first cycle of induction together with etoposide and daunorubicin (ADE), although this was not found to impact remission rate, EFS, or OS. Toxicity was generally similar in both arms of the trial, although patients treated on the high-dose arm had a greater incidence of serious fungal infections. The SJCRH trial incorporated MRD measurements during induction to guide therapy. MRD by MPFC was determined on a bone marrow sample obtained on day 22 of induction. Patients with MRD greater than 1% on day 22 received induction II before WBC count recovery. Patients with MRD greater than 1% at the end of the first month of induction I received GMTZ in addition to the second cycle of ADE. Patients with CNS-positive disease received triple intrathecal therapy. Patients who were MRD positive after induction I had a significantly worse outcome than patients who were MRD negative (3-year EFS, 43% vs. 74%). However, subgroup analysis revealed that this difference was only found in patients with high-risk AML at diagnosis ( FLT3 -ITD positive, monosomy 7, t(6;9), megakaryoblastic leukemia, therapy-related AML, or AML after MDS) or those with MRD levels greater than 1%. Low- and standard-risk AML patients or those with what was referred to in this study as low-level MRD (0.1% to 1%) did not demonstrate any outcome differences versus those with negative MRD. However, 20 patients who were MRD negative ultimately experienced relapse. In the current SJCRH study, AML08, patients are randomly assigned to receive high-dose cytarabine, etoposide, and daunorubicin versus clofarabine and cytarabine in induction I. Low-risk patients (those with CBF leukemia with negative MRD after induction I) receive 4 courses of chemotherapy, whereas those at high risk for relapse— FLT3 -ITD positive, monosomy 7, t(6;9), t(8;16), t(16;21), megakaryoblastic leukemia without t(1;22), treatment-related AML, AML after MDS, or MRD greater than 0.1% after induction II—receive two to three courses of chemotherapy followed by allogeneic HSCT. Standard-risk patients receive four courses of chemotherapy and are then eligible to receive one course of natural killer cell therapy if they have a killer cell immunoglobulin-like receptor (KIR)-mismatched haploidentical family member. Patients with FLT3 -ITD receive sorafenib after each course of chemotherapy (J. Rubnitz, personal communication).
The BFM (Berlin-Frankfurt-Münster) trial for de novo AML, BFM-2010, is for all comers, including patients with APL. Patients are being randomized to ADE ± GMTZ in induction I with MRD measurements on day 8, 15 and 28. Induction II includes cytarabine and idarubicin (AI) for patients with inv(16) and APL (who also receive ATRA). Other favorable-risk patients, including those with t(8;21), NPM1 mutations, and CEBPA , receive HAM (high-dose cytarabine 3 g/m 2 and mitoxantrone), and high-risk patients receive HAM plus targeted therapy, such as sorafenib if FLT3 -ITD positive, or dasatinib if KIT mutant positive. The third course is hAM (high-dose cytarabine at 1 g/m 2 ) for those who received AI and AI for low-risk patients who received HAM, with AI plus cladribine for the high-risk patients. Course 4 is hAM, except for the inv(16) and APL patients who receive cytarabine or cytarabine and etoposide as a final cycle. For all other patients, course 5 is high-dose cytarabine and etoposide. Liposomal daunorubicin, available in Europe, will be used in this trial. Prior studies have shown it to have equal efficacy in induction to idarubicin with less toxicity (G. Kaspars, D. Reinhardt, and D. Johnston, personal communication, January 2013).
Postremission Therapy
Several randomized studies have demonstrated that without postremission therapy almost all patients with newly diagnosed AML suffer a relapse within 2 years. The strategies for postremission therapy include autologous HSCT, allogeneic HSCT, or continued courses of chemotherapy. The CCG trials have shown no benefit to autologous HSCT over continued cycles of chemotherapy so, although that strategy continues to be used in some European trials, autologous HSCT is not included in current U.S. trials. Most studies have shown a lower relapse rate for AML patients treated with matched related donor HSCT versus chemotherapy, but the OS is often no better because of the significant morbidity and mortality of HSCT. For this reason, until the most recent trials, unrelated donor HSCT was believed to be too morbid for patients in first remission and thus trials used a genetic randomization, wherein all patients with a matched related donor proceeded to HSCT and all others proceeded with further cycles of chemotherapy.
Based on the success of the MRC trials with chemotherapy alone using a combination of cytogenetic characteristics and response to therapy, the COG and SJCRH adopted risk-based criteria for HSCT. HSCT, even from a matched related donor, is not recommended for patients with low-risk disease. This was previously defined by COG as the presence of t(8;21) or inv(16) in the leukemic clone at diagnosis, but more recently the presence of a CEBPA or NPM1 mutation has been added to this definition, based on the data revealing the better outcome for these patients. The COG has classified high-risk disease as monosomy 7, −5/5q−, FLT3 -ITD with high allelic ratio, MRD positive after induction chemotherapy, or more than 15% blasts in the marrow after one course of induction chemotherapy. * The incorporation of MRD after induction has enabled the COG to relegate normal karyotype patients to high- and low-risk groups, thereby dichotomizing patients and eliminating the intermediate-risk category. Therefore on the current AAML1031 protocol, high-risk patients will receive best available donor HSCT for consolidation, whereas good-risk patients will not go to transplant even if a sibling donor match is available. With recent advances in supportive care and improved therapy to prevent and treat graft-versus-host disease (GVHD), outcomes have improved for matched related and matched unrelated donor HSCT. Therefore for patients with high-risk disease, both trials recommend HSCT in first CR with an unrelated donor if no matched family donor is identified. The optimal number of cycles of chemotherapy before HSCT to balance minimizing toxicity while minimizing MRD is variable, with the MRC10 trial using four cycles, the current SJCRH trial including two to three cycles, and the COG trial using three cycles before HSCT.
* References .
For patients who do not proceed to HSCT there is controversy as to how many courses of chemotherapy are needed and what chemotherapy should be included. Several trials have shown that cycles of high-dose cytarabine after remission are associated with improved long-term survival. † MRC12 randomized patients to receive four versus five total cycles of chemotherapy, with early results suggesting that five cycles are superior; thus that strategy was subsequently used in the current U.S. trials. However, more recent data from the MRC have shown that the fifth cycle added toxicity with no additional therapeutic benefit. These findings are reflected in the current COG trial in which only four cycles of chemotherapy are used for good-risk patients with elimination of the high-dose cytarabine and asparaginase cycle (Capizzi II) that previously comprised the fifth cycle on AAML0531 and AAML03P1. High-risk patients for whom a donor is not identified also only receive four cycles of chemotherapy, but they do receive Capizzi II as their last consolidation cycle. The SJCRH trial has tried to make up for the lack of amsacrine by tailoring the first postinduction cycle based on the patient’s AML subtype and cytogenetics. On AML02, patients with t(9;11) or inv(16) AML receive cytarabine and cladribine, patients with M4/M5 AML receive cytarabine and etoposide, and patients with t(8;21) and all others receive high-dose cytarabine and mitoxantrone followed by two courses consisting of cytarabine and asparaginase followed by cytarabine and mitoxantrone. However, based on small subgroup numbers, AML08 uses a single backbone for all patients with the addition of sorafenib for patients with FLT3 -ITD (J. Rubnitz, personal communication, January 2013). CNS leukemia in patients with AML is highly sensitive to chemotherapy, and the addition of cranial irradiation does not decrease CNS relapse compared with systemic and intrathecal chemotherapy. Therefore, cranial irradiation is no longer part of the therapy for patients with AML. However, CNS-positive patients defined on the current COG AML study as having even one blast in the cerebrospinal fluid are treated with additional intrathecal cytarabine until the fluid is clear of blasts and for two additional intrathecal treatments beyond. Furthermore, in pediatrics, the addition of prolonged low-intensity maintenance therapy is associated with lower OS because of more resistant disease at relapse, so maintenance therapy is not used in the current U.S. trials.† References .
Supportive Therapy at Diagnosis and during Therapy
The diagnosis and treatment of childhood AML require a multidisciplinary approach involving pediatric oncologists, hematopathologists, pediatric surgeons, infectious disease specialists, pediatric intensivists, and pediatric nurses. While studies and procedures are initiated to make the diagnosis, special care must be taken to prevent and treat the life-threatening consequences of leukemia and accompanying pancytopenia. The most serious complications of leukemia at diagnosis are infection and bleeding with pancytopenia, tumor lysis syndrome, and leukostasis. The most common causes of death once remission is achieved are relapse, infection, hemorrhage, and cardiac events. The improvement in OS of patients with AML over the past several decades is attributable as much to better supportive care efforts as to more effective treatment regimens.
Infectious Complications.
At the time of initial presentation, up to 40% of patients with leukemia exhibit fever, and most of these patients are neutropenic or have impaired neutrophil function. During remission induction and subsequent cycles of chemotherapy, all AML patients experience profound prolonged neutropenia and are at high risk for life-threatening bacterial infections, particularly viridans group streptococcal infections. Almost all AML patients experience at least one febrile neutropenic episode during therapy, and approximately 40% of these patients will have a documented infection; most commonly these are bacterial infections, followed by viral infections and then fungal infections. Infections account for nearly two thirds of deaths in patients in remission. Therefore supportive care during induction and postremission therapy must pay particular attention to prophylactic, empiric, and treatment coverage for infectious complications. Several strategies are used to prevent infection or decrease mortality from infection. Some centers isolate patients receiving AML therapy in laminar airflow rooms during episodes of neutropenia. The supportive care guidelines for the CCG 2961 study included mandatory hospitalization for all patients, whether febrile or not, until the absolute phagocyte count had risen, and this rule has been strongly recommended in current COG trials. On CCG 2961, almost 50% of patients developed gram-positive infections and approximately 25% developed gram-negative infections across all phases of therapy. Infection-related mortality was 11%, including death from fungus. Bacterial agents included coagulase-negative staphylococci and α-hemolytic streptococci (including viridans streptococci) as well as Klebsiella and Pseudomonas species. To decrease the rate of bacterial infections, some programs routinely prescribe the use of gut decontamination, mouthwashes, or prophylactic antibiotics. A study by SJCRH demonstrated that initiation of antibiotics at the onset of neutropenia prevented sepsis, with the optimal regimen including both vancomycin and ciprofloxacin. This remains an area of controversy because, in some studies, although prophylaxis may decrease the rate of bacterial infection it may also be associated with an increase in drug-resistant organisms. Further investigation is indicated in a prospective randomized fashion before it is widely adopted. For example, in the SJCRH study, an earlier regimen of vancomycin and cefepime resulted in two cases of life-threatening resistant gram-negative bacteria and one of Bacillus cereus , leading to the change to ciprofloxacin for gram-negative coverage. The COG is currently conducting a study of a related fluoroquinolone, levofloxacin, for bacterial prophylaxis in AML and patients with relapsed ALL. Patients will be randomized to receive levofloxacin versus no bacterial prophylaxis from the initiation of chemotherapy for two consecutive cycles. In contrast to the controversy regarding bacterial prophylaxis, there is broad agreement that patients who are febrile at initial presentation and patients who experience febrile neutropenia during therapy should be immediately started empirically on broad-spectrum antibiotics, including vancomycin, once cultures have been obtained. In many centers, vancomycin is discontinued after 48 to 72 hours if there is no evidence of gram-positive bacteremia. Typhlitis, or generalized bacterial infection of the cecum, is more common in children than in adults and is more common in patients treated with doxorubicin instead of daunorubicin. Although this is a potentially life-threatening complication, conservative management with broad-spectrum antibiotics and nothing by mouth is generally preferable to surgical treatment in the setting of profound neutropenia.
AML patients are at high risk for invasive fungal infection, which carries a high risk for mortality. The routine use of fungal prophylaxis is becoming more routine given the intensity of AML protocols. Data from the CCG 2961 study demonstrated Candida and Aspergillus infections in approximately 13% of patients across all phases of therapy and a strong association with infection-related mortality. Some early studies have shown a decrease in some Candida species with fluconazole prophylaxis, but an increase in Candida krusei infections was documented, with no decrease in infection by invasive molds. Two meta-analyses have concluded that there is a reduction in morbidity because of invasive fungal infections when prophylaxis is used for those with prolonged neutropenia and post-HSCT patients. A randomized controlled trial of posaconazole versus fluconazole or itraconazole as fungal prophylaxis in neutropenic patients with AML or MDS has shown a lower rate of invasive fungal infections and improved OS in the posaconazole group. However, posaconazole has not been studied in children younger than age 13 years and, owing to exclusively oral administration, may have limitations during administration of emetogenic AML chemotherapy. Voriconazole has been studied as a prophylactic agent that has activity against both yeast and aspergillus, but the incidence of invasive fungal infections was no different compared with fluconazole or itraconazole. Voriconazole, similar to posaconazole, is metabolized through hepatic CYP450, leading to numerous interactions with other drugs and contributing further to a lack of enthusiasm in using either of these agents prophylactically. Thus fluconazole remains the most frequently used agent for fungal prophylaxis but lacks activity against Aspergillus . Currently the COG is conducting a study randomizing patients with AML to caspofungin versus fluconazole for fungal prophylaxis. Caspofungin is from the echinocandin family of antifungal agents that functions by targeting fungal cell wall biosynthesis of β-1,3-glucan. It is effective against both Candida and Aspergillus species and has safety data in children as young as 3 months of age. Caspofungin is given intravenously and is generally well tolerated but has minimal CNS penetration.
A test to detect galactomannan-containing Aspergillus antigens in the sera of immunocompromised patients has become commercially available. At this point, the galactomannan assay has been used most advantageously to follow the status of invasive aspergillosis and its response to therapy. However, some centers do not use fungal prophylaxis during AML therapy but rather screen twice weekly with the galactomannan assay and use a low threshold for computed tomography (CT) to detect early invasive disease. Whether or not fungal prophylaxis is used, empirical fungal therapy should be initiated for neutropenic patients who remain febrile for longer than 5 days, although some centers use a threshold of 72 hours. Patients who need empiric fungal therapy should be routinely scanned during neutropenia and after the recovery of circulating neutrophils be screened for evidence of fungal disease, and the diagnosis of fungal infection should be made by biopsy whenever feasible. Amphotericin has long been the antifungal agent of choice for empiric therapy, and the liposomal forms appear to be equivalent in efficacy and less toxic to renal function. One prospective randomized trial has shown that caspofungin as empiric fungal therapy is as effective as liposomal amphotericin B and is generally better tolerated. Whereas caspofungin is well tolerated with few drug interactions, as mentioned earlier, it does not cross the blood-brain barrier and thus is ineffective at treating suspected or proven CNS fungal infections. Voriconazole as empiric fungal therapy was compared with amphotericin in a prospective randomized trial and found to provide comparable coverage with less renal toxicity but with more visual changes and hallucinations. Voriconazole is seldom used empirically unless Aspergillus is highly suspected, such as in the case of prolonged febrile neutropenia with pulmonary lesions on imaging.
All patients undergoing AML therapy should receive prophylaxis against Pneumocystis carinii (now called P. jirovecii ) pneumonia (PCP). The first-line agent in PCP prophylaxis is trimethoprim-sulfamethoxazole, but in some patients this is associated with significant allergic reaction or prolonged bone marrow suppression. Dapsone is also effective as PCP prophylaxis in pediatric oncology patients but is associated with significant hemolysis and methemoglobinemia in some patients. Atovaquone has been shown to be highly effective as PCP prophylaxis for pediatric leukemia patients, although it may be associated with gastrointestinal side effects, including pancreatitis. Aerosolized pentamidine has also been shown to be effective as PCP prophylaxis in children with leukemia, although it may be associated with significant bronchospasm and can only be administered to children old enough to comply with the inhalational administration (usually >5 years).
Viral infections can be a significant cause of morbidity and mortality in AML patients as well. CCG 2961 and COG AAML0531 advocate intravenous immune serum globulin (IVIG) administration for low IgG levels, and some centers also recommend respiratory syncytial virus (RSV) prophylaxis for infants. All blood products transfused should be leukoreduced and cytomegalovirus (CMV) negative whenever possible. Acyclovir may be required for patients with recurrent zoster or mucocutaneous herpes simplex.
Growth factors, such as G-CSF and granulocyte-macrophage colony-stimulating factor (GM-CSF), have been used in the past, both as priming agents to synchronize the cell cycle before chemotherapy and as supportive care to decrease the duration of neutropenia after chemotherapy. Trials have shown that although G-CSF does shorten the duration of neutropenia its use does not lead to fewer episodes of febrile neutropenia, fewer microbiologically documented infections, less infection-related mortality, or improved OS. Based on these data, most centers no longer advocate the routine use of growth factors as supportive care during AML therapy.
Bleeding Complications.
Severe thrombocytopenia is not unusual in leukemia patients, and hemorrhage is one of the leading causes of early mortality in these patients. AML patients who present with bleeding and thrombocytopenia should be treated with platelet transfusions. In patients who are thrombocytopenic but do not have active bleeding, concern still remains about spontaneous bleeding, particularly intracranial bleeding, which can be catastrophic and have irreversible sequelae. The level of thrombocytopenia that warrants prophylactic platelet transfusion to prevent bleeding, however, is controversial. Some early observations suggested that platelet counts less than 20,000/µL predispose to increased bleeding, and that number became a standard threshold for transfusion. However, this was primarily in patients also receiving aspirin therapy, and subsequent studies have shown spontaneous bleeding occurs at a similar rate with platelet counts of less than 10,000 or 20,000/µL; and many centers have adopted 10,000/µL as the new threshold for transfusion. The threshold for platelet transfusion in patients undergoing procedures such as line placement or lumbar puncture has also not been well studied, and the accepted standard threshold of 50,000/µL may be higher than necessary. All blood products should be leukoreduced or CMV negative to prevent CMV infection, and irradiated to prevent transfusion-related GVHD. Menstruating women may require hormonal menstrual suppression for the duration of therapy with expected platelet drops to less than 50,000/µL.
Patients with AML, particularly APL, may also present with DIC, and coagulation studies should be a routine part of the initial workup in a patient with AML. In APL, overexpression of one of the plasminogen receptors, annexin A2, has traditionally been implicated as the cause of increased production of the fibrinolytic protein plasmin, which may be part of the cause of DIC in these patients. However, recent in vitro studies performed in APL cell lines suggest that, in fact, a different plasminogen receptor, S100A10 (also known as p11), may actually be the culprit. Levels of S100A10 increased concomitantly with PML-RARA induction, and targeted inhibition of SA100A10 expression resulted in decreased fibrinolytic activity. Patients whose leukemic cells are successfully treated will exhibit resolution of DIC with the reduction in blast cells but, while the blast count is decreasing, DIC should be treated with fresh-frozen plasma, cryoprecipitate, or recombinant factor VIIa as needed to prevent bleeding. For patients with APL, the initiation of ATRA before chemotherapy leads to rapid resolution of DIC and has markedly reduced the early mortality previously associated with APL. Since the development of ATRA, patients with APL are no longer routinely treated with low-dose heparin to prevent DIC.
Complications of Hyperleukocytosis.
Patients with AML, particularly those with high presenting leukocyte counts, have an increased risk for tumor lysis syndrome. In patients with leukemia, the rate of blast cell turnover can exceed the body’s ability to metabolize and excrete the contents of dying cells, leading to life-threatening metabolic derangements. Hyperuricemia resulting from the metabolism of excessive amounts of released nucleic acids can cause renal failure. The ensuing hyperkalemia may lead to fatal cardiac arrhythmias, and hyperphosphatemia and hypocalcemia may cause seizures and worsen the renal failure. Elevated blood urea nitrogen levels cause platelet dysfunction, which may exacerbate an existing coagulopathy. Vigorous intravenous hydration and administration of bicarbonate and other agents, such as allopurinol or recombinant urate oxidase (rasburicase), should be started immediately to decrease formation and increase the solubility and renal excretion of insoluble urate. Recombinant urate oxidase can dramatically decrease uric acid levels within a few hours but, in patients with G6PD deficiency, it can cause methemoglobinemia and life-threatening hemolysis.
Patients with WBC counts higher than 100,000/µL have an increased risk for leukostasis. Leukostasis causes intravascular clumping of blasts, leading to sluggish blood flow and subsequent hypoxia, hemorrhage, and infarction of tissue. Leukostasis can be life threatening when it occurs in the organs at greatest risk, the brain and lungs. Patients with pulmonary leukostasis present with significant hypoxia and tachypnea, which may progress rapidly to respiratory failure. CNS leukostasis may produce symptoms of confusion, headache, and somnolence and can lead to coma, stroke, and death. Hydration and hydroxyurea are therapies that can be initiated immediately to decrease the blast count. Leukapheresis or exchange transfusion can reduce the number of circulating blasts quickly, although this effect is transient, and therapy with cytoreductive drugs should be started as soon as possible. Routine pheresis for patients with a WBC count higher than 100,000/µL has resulted in lower mortality rates during induction compared with historic controls. In one study, patients who achieved a higher than 30% decrease in total WBCs through pheresis had a higher CR rate than those who achieved less than a 30% decrease. Other retrospective reviews have shown a decrease in early mortality after leukapheresis but not an improvement in OS. Cytoreductive chemotherapy should be initiated as rapidly as possible because other measures to reduce leukemic blast numbers have only temporary effectiveness.
Chemotherapy Complications.
High-dose cytarabine is associated with chemical conjunctivitis, and dexamethasone or prednisolone eye drops were traditionally used to prevent this side effect. However, recently owing to the unavailability of this steroid formulation, artificial tear products have been effectively substituted. High-dose cytarabine is also associated with neurotoxicity, including seizures and cerebellar dysfunction, which may not be reversible. Patients should be monitored with frequent neurologic examinations, and high-dose cytarabine should be immediately discontinued if cerebellar signs develop. Cytarabine may also be associated with an impressive febrile response, which is difficult to distinguish from an infection-related fever. AML chemotherapy is associated with significant nausea and vomiting, and aggressive antiemetic regimens should be instituted. However, because of the high risk for fungal infection in these patients, dexamethasone should be avoided as an antiemetic. Mucositis is a significant toxicity during AML therapy, predominantly due to anthracyclines, and almost all patients require parenteral pain medication and nutrition. Malnutrition at the time of diagnosis appears to carry a worse prognosis in children with leukemia and, even in those who are well-nourished at diagnosis, weight loss during chemotherapy should be monitored closely, followed by the initiation of parenteral or enteral feedings in those who lose more than 10% of their initial body weight.
Renal toxicity during AML therapy is common and may be caused by a combination of chemotherapy, tumor lysis syndrome, sepsis, or treatment with antibacterial, antifungal, and antiviral agents. A creatinine clearance or glomerular filtration rate should be checked in patients with abnormal creatinine clearance or prolonged use of medications with renal toxicity. For patients with a creatinine clearance lower than 60 mL/min/1.73 m 2 , high-dose cytarabine, etoposide, and other renally cleared chemotherapy agents are usually given at reduced dosages. Significant liver toxicity can also be observed during therapy for AML, particularly for regimens containing the humanized anti-CD33 monoclonal antibody, GMTZ. GMTZ has been associated with sinusoidal obstructive syndrome and veno-occlusive disease, most commonly when used as a component of combination therapy or in patients who are heavily pretreated or who have undergone prior HSCT or undergo subsequent HSCT within 3 months of treatment with GMTZ.
AML regimens rely heavily on anthracyclines and their derivatives. The cumulative anthracycline dose in these regimens is often 300 mg/m 2 or higher in daunorubicin equivalents and therefore carries a high risk for cardiac toxicity. Cardiac deaths are one of the top three causes of death in remission for AML patients. All patients should undergo cardiac assessment before administration of chemotherapy cycles containing anthracyclines and yearly after chemotherapy is complete, until growth is completed in adulthood. However, recent pharmacogenomic studies suggest that certain genotypes, such as the rs7853758 variant within the SLC28A3 solute transporter, may particularly predispose children to developing anthracycline-induced cardiotoxicity. In vitro evidence has suggested that the cardioprotectant dexrazoxane works synergistically, not antagonistically, with anthracyclines against AML blasts, and there is increasing interest in incorporating this medication into AML clinical trials. On the current COG AML trial, use of dexrazoxane is permitted at the discretion of the treating physician and data will be collected to determine the impact on both cardiac outcome and secondary malignancy.
Extramedullary Disease
Between 20% and 40% of children with AML have evidence of extramedullary disease at the time of diagnosis. The most common manifestations of extramedullary disease are skin infiltrates, soft tissue or bone involvement, gingival infiltration, and CNS involvement. Some patients, particularly infants, may have isolated extramedullary leukemia without other evidence of disease, but bone marrow infiltration invariably occurs in these patients without systemic therapy.
CNS Involvement.
CNS involvement is more common in AML patients with high presenting WBC counts, favorable cytogenetic changes—t(9;11), t(8;21), and inv(16)—and children younger than 2 years. One recent retrospective review of almost 1500 children enrolled on COG AML trials found an incidence of CNS disease (defined as CNS3 status >5 WBCs per high power field with the presence of blasts) of 11%. AML patients with CNS disease at diagnosis have a higher risk for CNS relapse, but they often have a better overall prognosis because CNS disease is more common in the favorable-risk cytogenetic groups, which carry a lower risk for systemic relapse. Specific CNS-directed therapy in AML may also be less important than in ALL because several AML chemotherapy agents, including high-dose cytarabine, have excellent CNS penetrance. Cranial radiation was previously used in AML patients as prophylaxis against CNS relapse, but because of concerns about the myriad side effects of CNS radiation, including secondary malignancy, endocrine and growth impairment, and neurocognitive toxicity, later trials attempted to minimize radiation. In the BFM-87 study, all patients received intrathecal cytarabine, but the low-risk patients were randomized to additional therapy with cranial radiation or no further CNS treatment. An interim analysis demonstrated no increase in CNS relapse in the group that did not receive cranial radiation, so the radiation arm of the trial was closed early. CNS radiation therapy is no longer routinely part of AML treatment protocols. Intrathecal chemotherapy with cytarabine alone, methotrexate alone, or triple intrathecal therapy with cytarabine, methotrexate, and hydrocortisone have been reported to be effective as CNS treatment and prophylaxis. As prophylaxis for CNS-negative patients, COG study AAML0531 used intrathecal cytarabine alone at the start of each cycle, except for the Capizzi II cycle, which includes 3 g/m 2 of systemic cytarabine. For patients who had CNS disease at diagnosis, twice-weekly intrathecal therapy with cytarabine alone was used until the cerebrospinal fluid was clear, plus an additional two doses, with a minimum of four doses and a maximum of six doses. If more than six doses were required, the patient was taken off study for refractory CNS disease. These guidelines have been maintained on the current AAML1031 study. The SJCRH AML02 trial used a prophylaxis regimen of triple intrathecal chemotherapy with cytarabine, methotrexate, and hydrocortisone given at the start of each cycle but separated by at least 24 hours from systemic cytarabine dosages greater than or equal to 1 g/m 2 to prevent neurotoxicity. Patients with CNS disease at diagnosis receive once-weekly triple intrathecal chemotherapy for four doses. To decrease mucosal toxicity from the methotrexate, all patients receive leucovorin rescue at 24 and 30 hours after the intrathecal administration.
CNS relapse occurs in 2% to 8.8% of patients with AML. Most AML patients who suffer a CNS relapse will simultaneously or soon thereafter develop a bone marrow relapse. Truly isolated CNS relapse is rare and is associated with age younger than 2 years, hepatosplenomegaly at diagnosis, elevated presenting WBC count, M5 AML, and chromosome 11 abnormalities. For those with a truly isolated CNS relapse, the optimum treatment plan is unclear. There have been survivors reported after treatment with intrathecal chemotherapy alone, combined intrathecal and systemic chemotherapy, radiation therapy, and HSCT.
Chloromas.
Extramedullary infiltrations of the soft tissue, also known as chloromas, myelosarcomas, or granulocytic sarcomas, occur in 4% to 5% of children with AML. They may also be documented in patients with CML, MDS, or rarely in ALL and may be initially confused with non-Hodgkin lymphoma. Chloromas most often present simultaneously with AML or shortly before the systemic evidence of disease, but they may be isolated. The prognostic significance of extramedullary disease is not clear. Some groups have reported an unfavorable prognosis associated with chloromas, but others have demonstrated a more favorable outcome. Blast cells from patients with chloromas often exhibit the relatively favorable t(8;21). Most studies from Europe, Saudi Arabia, Japan, and the United States have reported no influence of chloromas on the OS of children with AML, and a recent review of four COG studies in fact demonstrated that children with orbital or CNS-based chloromas had significantly higher CR rates and OS compared with counterparts without chloromas treated on these same studies. Accordingly, most centers treat patients who have chloromas with standard AML regimens, because there is no consistent evidence supporting more intensive therapy. A beneficial role for radiation therapy in the treatment of chloromas has not been demonstrated. In a CCG trial, patients with chloromas were treated with local irradiation in addition to systemic therapy. There was no difference in EFS or in the incidence of local recurrence compared with the chemotherapy-only regimen. However, chloromas arising in the orbit or CNS may cause loss of vision or cord compression, and emergency radiation therapy as well as chemotherapy is indicated in these situations.
Prognosis
Although historically based exclusively on morphologic subtype, the ability to predict prognosis for patients with AML in the current genomic era derives from a composite of cytogenetic features, molecular subtype, and response to initial chemotherapy as evaluated by MRD. The recent integration of these features on the background of more intensive therapy with better supportive care has enabled a gradual improvement in pediatric AML outcome ( Fig. 51-6 ). Classically, FAB subtype M0, which commonly harbors a complex karyotype, abnormalities of 5 or 7, or trisomy 8, is associated with a poor prognosis. M7 AML (non–Down syndrome), often associated with monosomy 7, carries a poor prognosis. M3 AML with the t(15;17) a group previously associated with a dismal prognosis, has emerged as a highly favorable subtype owing to responses with the addition of ATRA and As 2 O 3 to chemotherapy regimens. Similarly, CBF AML with t(8;21) or inv(16) and normal karyotype AML with either an isolated CEBPA or NPM1 mutations are favorable subtypes. By contrast, FLT3 -ITD mutations with high allelic ratios are associated with a poor prognosis, as are patients without good-risk molecular features who have significant residual MRD after induction therapy.
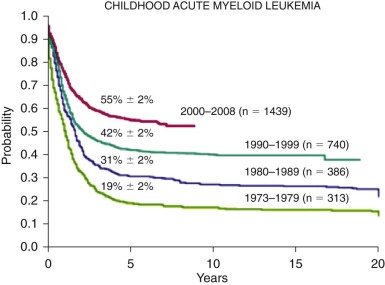
Demographic Risk Factors
Age older than 10 years was historically reported to carry a worse prognosis in clinical trials at SJRCH, as well as from the BFM and Japanese groups. However, a recent report from the SJCRH AML02 study demonstrated comparable EFS and OS in children as old as age 21 years as in their younger counterparts, although these older children had higher toxic death rates predominantly of infectious etiology. Race and ethnicity play a role in prognosis as well. Patients on CCG2891 who received chemotherapy alone had an OS of 48% for white children versus only 37% for Hispanic children ( P = .016) and 34% for black children ( P = .007). On the SJCRH AML97, there was a trend toward inferior outcomes for black compared with white children (hazard ratio, 1.61; 95% confidence interval, 0.76 to 3.42; P = .22 with 5-year survival rates of 55.6% vs. 27.3%). In these trials, there was no difference in other prognostic variables between the groups and these patients were hospitalized and received intravenous chemotherapy, received the same supportive care, and had the same compliance rates. Therefore, the observed differences may be attributable to pharmacogenomic differences that affect response to and toxicity from chemotherapy. Patients who are overweight (body mass index ≥95th percentile) or underweight (≤10th percentile) also carry a worse prognosis and are less likely to survive and more likely to suffer treatment-related mortality.
Cytogenetic Factors
Results of several large multicenter trials have confirmed the correlation between certain cytogenetic abnormalities and clinical outcome and have shown that karyotype in AML represents an independent prognostic variable for attainment of complete response and OS. In children and adults, the highest complete response rates and longest survival times have been linked to association with t(8;21) and inv(16) or t(16;16). These translocations result in modifications to the core-binding complex, a group of proteins that appear to play a vital role in hematopoiesis. AML cells with inv(16) have increased incorporation of cytarabine into nuclear DNA and are more susceptible to cytarabine chemotherapy in vitro. Patients with APL and t(15;17) previously represented a poor-risk group, but, since the advent of ATRA and As 2 O 3 therapy (see later), now represent a group with a favorable prognosis. Other cytogenetic aberrations have been associated with an unfavorable outcome. These include monosomy 5, del(5q), monosomy 7, abnormalities of 3q, aberrations of 12p, and complex cytogenetic abnormalities. For pediatric patients with monosomy 7, several studies have shown better OS when HSCT is used as consolidation therapy as opposed to conventional chemotherapy. Patients with normal karyotypes have traditionally been grouped together in an intermediate-prognosis group, but now mutations such as the FLT3 -ITD (poor prognosis) and NPM1 and CEBPA (good prognosis) have been demonstrated to alter prognosis sufficiently to influence the risk assignment of these patients at diagnosis on COG, SJCRH, BFM, and MRC trials. Abnormal karyotypes linked with an intermediate prognosis include del(7q), 11q23 translocations, +21, +8, +22, and del(9q). Other karyotypes such as t(6;9)(p23;q34), t(8;16)(p11;p13), and t(16;21)(q24;q22) are uncommon but appear associated with a poorer prognosis. While more studies with large numbers of patients undergoing cytogenetic evaluation are completed, certain groups of karyotype aberrations are showing heterogeneity in their prognostic significance. For example, all 11q23 translocations were previously grouped together in the intermediate- to poor-prognosis category. However, studies have shown that the 11q23 aberration may be subdivided into groups of lesser and greater prognostic risk. Patients with the t(9;11)(p22;q23) have been found in some studies to have a longer EFS and OS than patients with other 11q23 translocations. However, this was not found to be true in a recent review of outcome on MRC10 and 12 trials. As new therapeutic agents are developed that target the specific molecular aberrations identified by cytogenetic analysis, accurate detection of these abnormalities becomes even more important.
Molecular Genetic Factors
Analysis of molecular genetics is emerging as an invaluable tool in prognostic determination for patients with AML, as well as for providing insight into the pathogenesis of AML and elucidating possibilities for targeted therapy. RAS mutations have been identified in pediatric AML patients, with NRAS mutations in 10% to 15% and KRAS mutations in 3% of patients. HRAS mutations are rare in AML. In both adults and children these mutations are associated with favorable-risk AML. In adults, the association is with CBF leukemias and, in particular, inv(16). Although some pediatric studies have also found a link between CBF AML and NRAS mutations, others have found a distinct absence of NRAS with inv(16) and a unique correlation of NRAS and NPM mutations not seen in adult AML. Independently, however, NRAS mutations have no significant impact on prognosis. Patients had similar 5-year EFS and OS, although patients with NRAS mutations were associated with increased treatment-related mortality on one study. KIT mutations have been identified in 11.3% of pediatric AML patients, predominantly in the CBF leukemias, ranging from 17% to 41% of patients. The role of KIT mutations in prognosis has been unclear: in some series, the KIT mutations do not seem to have prognostic significance, in others, KIT mutations have been shown to confer worse prognosis in patients with t(8;21) but not in patients with inv(16) and, in still other trials, KIT mutations confer a worse prognosis in all subsets of CBF leukemias. However, a recent review of more than 200 children with CBF AML enrolled on four COG AML trials concluded that KIT mutations do not alter prognosis. Owing to their good outcome with chemotherapy alone, patients with CBF AML are currently nonrandomly assigned to chemotherapy without HSCT in CR1 even if a matched family donor is available. Based on these recent data, on current COG trials the presence of a KIT mutation does not alter risk stratification. This is in contrast to adult data which show that KIT mutations appear to confer an inferior outcome. These prognostic differences in adult versus pediatric CBF AML may reflect more aggressive therapy in children and, potentially, an earlier cell of origin in the adult disease that contributes to increased resistance to therapy.
As noted, FLT3 mutations include ITDs in the juxtamembrane domain and point mutations in the activation loop (ALM). FLT3 -ITDs have a worse prognosis, particularly when present in a high allelic ratio (>0.4), whereas FLT3 -ALMs seem to have no impact on prognosis. Partial tandem duplications of the MLL gene have also been reported in 13% of pediatric AML patients and are associated with a worse prognosis in adult and pediatric patients. Frameshift mutations in NPM1 lead to cytoplasmic localization of nucleophosmin and have been identified in 6% to 8% of all children with AML and 25% to 30% of pediatric AML patients with normal cytogenetics. When identified as an isolated mutation in adults and children, it is associated with improved prognosis, but when found in conjunction with an FLT3 -ITD, the poor prognostic impact of the FLT -ITD prevails. With contemporary chemotherapy, children with AML with a normal karyotype and isolated NPM1 mutation have outcomes in keeping with CBF leukemia with OS greater than 80%; thus these children have been assigned to the low-risk arm on the current COG trial (R. Aplenc, personal communication, June 2011). PTPN11 mutations are found in only 4% of pediatric patients with AML (4%), and 18% to 36% of the PTPN11 mutations are seen in M5 AML. In pediatrics, this mutation does not appear to have prognostic importance. The Wilms tumor gene (WT1) is aberrantly expressed in AML; in adults, high expression levels of WT1 at diagnosis have been associated with a poor prognosis in many studies. In pediatric patients, however, the available data have been contradictory. One small study of 47 patients has shown that low levels of WT1 at diagnosis are associated with good outcome, whereas another small study of 41 patients has shown that high levels of WT1 are more commonly seen in association with the t(8;21) and inv(16) favorable prognosis cytogenetic findings, and therefore associated with a better prognosis. Hollink and colleagues found an association between WT1 mutations and an inferior outcome. More recently, a large study performed by the COG identifying 70 patients with WT1 mutations found that although there was significant overlap of the WT1 mutation with either CBF or FLT3 -ITD, independently, WT1 mutations did not influence prognosis. Interestingly, 28% of pediatric AML patients harbor a minor single nucleotide polymorphism (SNP) in exon 7 of WT1 (rs16754) that was found to result in an improved 5-year OS (62% vs. 44%) for patients treated on CCG 2961 compared with those with leukemic expression of the major WT1 allele. Levels of expression of wild-type WT1 may also influence prognosis. However, at present, WT1 mutational status, presence of the minor SNP, and wild-type expression levels continue to be studied but are not being used to make treatment decisions. In addition to its role as a partner in the t(8;21) translocation, RUNX1 may also be mutated in approximately 5% of pediatric AML patients, specifically with point mutations in exons 3 and 8. Although typically associated with AML secondary to MDS, in de novo AML these patients had less organomegaly, a higher incidence of chloromas, and a trend toward improved outcome with modern intensive AML therapy. However, RUNX1 mutations are not currently used for risk stratification.
CEBPA mutations have been found to be an independent prognostic marker conferring a good outcome in both pediatric and adult AML and, similar to that seen for CBF AML, at least in children. Interestingly, pediatric patients with AML and CEBPA mutations tend to be older (age >10 years) and predominantly have a normal karyotype with an absence of other good- or poor-risk molecular abnormalities. Furthermore, patients with CEBPA mutations did not benefit from HSCT in first remission. Thus, in current studies, presence of a CEBPA mutation classifies a patient as low risk. Recent next-generation sequencing–based studies have revealed frequent mutations in the DNA methyltransferase gene, DNMT3A, the gene coding for the demethylating enzyme, TET2, and the genes IDH1 and IDH2 , which code for proteins in the Krebs cycle. DNMT3A mutations are generally associated with a poor outcome and contribute to a worse prognosis when combined with FLT3 ITD. TET2 mutations are similarly associated with a poor prognosis, with or without corresponding FLT3 abnormalities. By contrast, IDH1/IDH2 mutations appear to be more positive prognostic features. However, these abnormalities are generally rare in pediatric AML ; thus their impact cannot be easily determined, and at this time they have been excluded from any current pediatric AML risk stratification schemas.
Minimal Residual Disease
Early response to treatment, as assessed by MRD in the bone marrow and/or the peripheral blood after induction therapy, has been shown to be an independent and powerful prognostic factor in pediatric leukemia. MRD can be detected by molecular means such as PCR assay, which is beneficial for the monitoring of leukemias with fusion genes or recurrent mutations. Alternatively, the use of multiparameter flow cytometry (MPFC) is more universally applicable, but it may be user dependent and vary significantly based on technical aspects, threshold levels, and timing of evaluation.
PCR-based MRD evaluation requires that a cytogenetic abnormality or mutation present at diagnosis will be predictive of relapse or refractory disease. In CBF AML, persistence of the AML1-ETO t(8:21) or CBFB-MYH11 inv(16) transcripts long after completion of therapy has challenged the value of following these lesions either during or after completion of therapy. However, a recent adult study has suggested that at least in AML with inv(16), PCR analysis may have more predictive power. Negative bone marrow results of PCR assay for the CBFB-MYH11 transcript (<10 detectable copies per cell) early in consolidation with two subsequent negative PCR evaluations either in the bone marrow or peripheral blood later in consolidation and within 3 months of completing therapy were highly predictive of remaining in CR, with a high correlation between bone marrow and peripheral blood measurements during surveillance. In APL and CML (see later), levels of PML-RARA or BCR-ABL transcripts have been found to be extremely valuable for monitoring, although the site of monitoring has been somewhat controversial. Standardized monitoring for BCR-ABL levels in the peripheral blood is now accepted as the standard of care in CML, whereas the recently closed COG trial in APL has mandated bone marrow evaluations during maintenance and subsequent follow-up. A number of adult and pediatric studies have examined following FLT3, CEBPA, NPM1, MLL rearrangements/PTDs, or WT1 mutations in those patients harboring these abnormalities at diagnosis. However, the utility of any of these mutations as a molecular MRD marker remains uncertain owing to stability of the mutation ( FLT3 -ITD) or normal background levels (WT1) . Thus, although their presence at diagnosis may be indicative of response, persistence of these lesions is not being routinely used to evaluate disease response in pediatric AML.
By contrast, use of MRD by MPFC has proven to be a more broadly applicable component of risk stratification in both pediatric ALL and AML. Particularly in the large subset of intermediate-risk AML, MRD has allowed the distinction of standard- and high-risk groups, whereas the use of morphology alone proved insufficient. The ability of MPFC MRD measurement to contribute to the evaluation of early treatment response and improve prognostic evaluation in pediatric AML was demonstrated for CCG 2961. OS for patients in morphologic remission after induction I was 41% if MRD was positive, compared with 69% for those who were MRD negative. A strong impact of MRD was also apparent in AML patients who were in morphologic remission at the end of induction I on the recent AAML03P1 study. The 46 MRD-positive patients of the 188 patients deemed in CR showed an increase in relapse risk (RR) at 3 years (60%, P <.001) and lower DFS (30%, P <.001) and OS (56%, P = .002) compared with those who were MRD negative (3-year RR, 29%; DFS, 65%; OS, 80%) ( Fig. 51-7, A ). Not only did this study demonstrate the adverse prognostic value of positive MRD compared with morphology alone but, conversely, showed that 26% of patients deemed as having disease based on morphologic bone marrow evaluation were in fact MRD negative and had a good outcome. Similar findings were identified by the SJCRH, confirming that recovering normal marrow blasts and leukemic myeloblasts can be challenging to distinguish morphologically even by the most experienced hematopathologist. Multivariate analyses on COG and SJCRH studies confirmed positive MRD as an independent adverse prognostic factor, although the SJCRH study found that this was only significant for MRD levels greater than 1% after induction I, or greater than 0.1% after induction II, whereas this distinction was not specifically identified in the COG studies. Subsequent analysis by the SJCRH demonstrated that an MRD threshold of 0.1% was independently predictive of outcome both after induction I and induction II (see Fig. 51-7, B ). The impact of MRD was independently confirmed in recent MRC and Dutch trials. Furthermore, COG studies found that MRD levels held their prognostic significance in normal karyotype AML. Based on these data, the COG has used MRD by MPFC to eliminate the intermediate-risk group of patients without prognostic cytogenetic or molecular features. Those patients with residual MRD at a threshold of 0.1% after the first induction cycle are deemed at high risk and relegated to more intensive therapy, whereas those with MRD levels below 0.1% are allocated to a low-risk study arm.
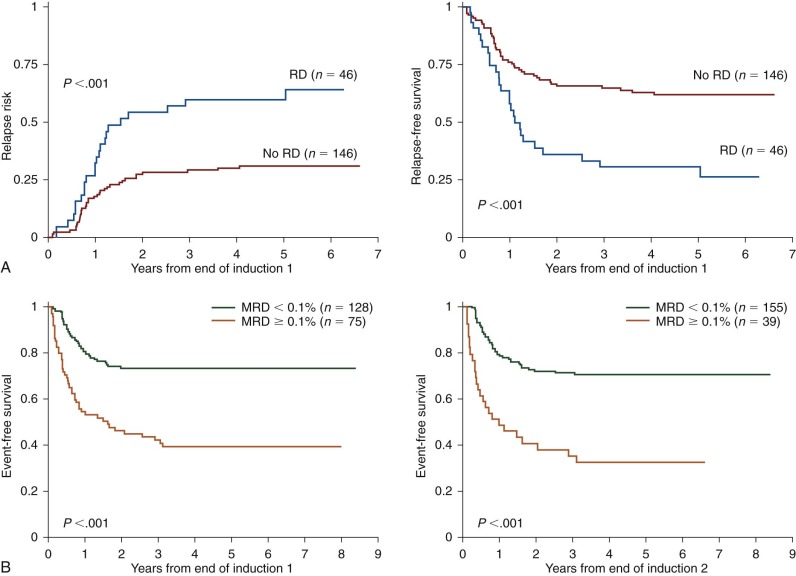
Relapsed or Refractory Disease
The prognosis is grave for children with disease refractory to induction chemotherapy or who have a disease relapse. Although 80% to 90% of pediatric patients with AML will achieve remission, 30% to 40% will later recur, and the OS remains only 40% to 60%. The bone marrow is the most common site of relapse in AML, and isolated CNS relapse is an uncommon finding, but isolated CNS relapse has been associated with prior CNS involvement.
Prognosis in Relapsed or Refractory Disease
A durable remission is less likely to be achieved and maintained in patients with relapsed or refractory AML than in patients with newly diagnosed AML, but the salvage rate in these patients has improved in recent years. The single most important prognostic indicator after relapse is duration of first remission, with less than 1 year to relapse carrying an OS rate of 0% to 21% and longer than 1 year to relapse carrying an OS rate of 33% to 62%. Other characteristics that have been associated with better prognosis after relapse are male gender, good-risk cytogenetic features of t(8;21) and inv(16), chemotherapy alone without HSCT as consolidation during first CR, and the use of HSCT as part of relapse therapy. The characteristics associated with worse prognosis after relapse are early recurrence, M5 or M7 morphology, poor-risk cytogenetics, and HSCT during first CR.
Conventional Therapy for Relapsed or Refractory Disease
There is no accepted standard therapy for adult or pediatric patients with relapsed or refractory AML. However, several researchers have demonstrated activity of high-dose cytarabine in patients with relapsed disease, even in those who had been exposed to cytarabine during prior treatment. High-dose cytarabine has been used in combination with other agents, such as mitoxantrone, asparaginase, etoposide, fludarabine, cladribine, and clofarabine. The combination of cytarabine and mitoxantrone was particularly effective as reinduction with a remission rate of 76%, but now that this regimen has been incorporated into the current standard first-line regimens, its use as a reinduction regimen may provide too much cumulative anthracycline for many patients. Fludarabine, cladribine, and clofarabine are all deoxyadenosine analogues that have been used for the treatment of AML. Trials using high-dose cytarabine in combination with fludarabine and G-CSF (i.e., the FLAG regimen) have demonstrated a 70% remission rate in patients with relapsed AML, and this regimen is frequently used by many centers for patients who have experienced a relapse of their disease. The addition of idarubicin to the FLAG regimen has resulted in longer-lasting remissions in some studies, but it has also been associated with more significant toxicity and for this reason is not frequently incorporated. The combination of cytarabine and cladribine has been successful as a relapse regimen in some trials for adult patients and pediatric patients with de novo AML, particularly those with the M5 subtype. However, this combination seems to be of little benefit in pediatric patients with relapsed or refractory AML. Clofarabine was found to be effective as a single agent in relapsed ALL and AML and was subsequently approved by the FDA for use in relapsed ALL. Clofarabine has been shown to be safe and effective in combination with cytarabine in regimens for adult patients, and this combination has been tested for pediatric patients with relapsed or refractory AML. The recently closed COG study AAML0523 examined the combination of clofarabine and cytarabine for relapsed AML and ALL and found that a dose of 52 mg/m 2 of clofarabine was tolerated by patients with relapsed AML. Of the 46 patients who were evaluable, only 21% responded as evaluated by complete response or complete response with incomplete platelet recovery, which did not meet the predetermined statistical threshold for efficacy in overall response rate. Based on these findings, this regimen has somewhat fallen out of favor compared with FLAG. However, as a result of compliance issues and failure to recognize complete response with incomplete count recovery as evidence of response, results from this study may in fact underestimate the efficacy of this combination therapy; thus clofarabine and cytarabine for relapsed AML as a bridge to HSCT (see later) may still be worthy of consideration. For patients who cannot tolerate high-dose cytarabine or further anthracyclines, one alternative regimen that has been used at the Memorial Sloan-Kettering Cancer Center is topotecan, vinorelbine, thiotepa, dexamethasone, and gemcitabine (TVTG), with 5 of 9 AML patients responding to this therapy.
Long-term remission for patients with relapsed or refractory disease requires consolidation therapy with allogeneic HSCT. Traditionally, this has included matched family members or matched unrelated donors, but the more recent use of alternative HSC sources, such as banked umbilical cord blood or haploidentical family donors, has increased the number of patients with a potential marrow source. A recent trend has been in using non–total-body irradiation (TBI)–based conditioning regimens for AML in first remission due to equal efficacy and less toxicity, but TBI may still be used in the conditioning of patients with relapsed AML. It is possible to cure patients who have persistent, refractory disease at the time of allogeneic HSCT, particularly if they have less than 20% blasts in the marrow. Those patients with shorter remissions and higher blast percentages in their marrow have traditionally been associated with only a 25% chance of cure, whereas for patients who have had a long first remission and proceed to HSCT in a second CR the OS rates are much better and may be as high as 40% to 62%. However, although lower levels of MRD were associated with improved overall outcomes, investigators at the SJCRH found that with contemporary risk-adapted therapeutic regimens, allogeneic HSCT may be curative for patients with AML who continue to be MRD positive or even have persistent marrow blasts, with 5-year OS of 67% and 58%, respectively. This analysis included patients with relapsed AML. These findings are in contrast to the impact of residual disease in ALL, which may be attributed at least in part to the graft-versus-leukemia (GVL) in AML. This effect is difficult to measure directly but has been indirectly inferred from the presence of GVHD. The presence of acute and chronic GVHD has been associated with lower relapse rates in AML. Donor lymphocyte infusions have been used successfully to induce GVL effect in patients with CML who experience relapse after allogeneic HSCT, but this strategy is much less effective in patients with AML who experience relapse after allogeneic HSCT.
There can be GVL effect without GVHD, however, as evidenced by lower relapse rates in patients receiving HSCT from a matched sibling versus an identical twin, even in the absence of GVHD. There is much interest in maximizing the GVL effect, and recent studies have concentrated on the role of alloreactive natural killer (NK) cells in this process. Donors who are mismatched at the KIR ligand produce NK cells that recognize and destroy host AML cells in response to the lack of expression of a self class I ligand on the AML cells. Deliberate KIR mismatching through haploidentical HSCT has been shown in small studies to decrease the relapse rate without increasing GVHD, therefore resulting in improved long-term survival. This strategy is being used in clinical trials for patients with high-risk de novo AML or relapsed and refractory disease and is currently incorporated into ongoing pediatric trials of the COG and SJCRH. The feasibility of engrafting haploidentical NK cells in children with AML in remission was recently demonstrated.
Novel Agents and Targeted Therapy
Conventional therapy for AML is predominantly a “shotgun” approach in which leukemia cells, as well as normal hematopoietic stem and progenitor cells and many other normal tissues, are injured, with the hope that the normal cells will recover faster and more completely than the leukemia cells. As discussed earlier, this approach has not resulted in superior cure rates in AML and the intensity has been increased to the point that most patients suffer toxicity and treatment-related morbidity; a substantial fraction of patients suffer treatment-related mortality. While the molecular pathogenesis of AML has been elucidated, opportunities have appeared for the development of novel and targeted agents that preferentially destroy leukemia cells, with far less significant effects on normal hematopoietic cells and other normal tissues. However, preclinical and adult clinical data, not to mention drug availability, are in rapid flux, given the growing interest in developing novel targeted therapeutics. Thus current therapeutic platforms for relapsed AML are moving to the model of having a standardized backbone to which novel agents can be rapidly inserted to be sufficiently nimble to adapt to the availability and promise of emerging drugs.
Antibody Therapy with Gemtuzumab Ozogamicin.
Gemtuzumab ozogamicin (GMTZ, also discussed earlier in the induction chemotherapy section) is a recombinant, humanized anti-CD33 monoclonal antibody linked to the tumor antibiotic calicheamicin. CD33 is expressed on most AML cells but not on pluripotent HSCs or other tissues, thus making it possible to target leukemia cells more specifically. GMTZ has been used successfully as a single agent for adults and children in relapse, with infusional side effects of fever and chills, hematologic toxicity resulting from pancytopenia, including some cases of prolonged thrombocytopenia, and hepatic toxicity ranging from transient liver function abnormalities to fatal venoocclusive disease, particularly when used within 3 months of HSCT. GMTZ as a single agent is not a particularly strong reinduction regimen, but it has now been used successfully in combination with cytarabine-mitoxantrone and cytarabine-asparaginase as part of a reinduction regimen. Given the data from the United Kingdom on the limited efficacy of GMTZ in initial adult AML therapy, current and future availability of this agent may be very limited. GMTZ may have a role specifically in relapsed APL, however (see later).
Receptor Tyrosine Kinase Inhibitors.
FLT3 (discussed in detail earlier) is a receptor tyrosine kinase that is upregulated in the AML cells of most patients. There are also two types of activating mutations in FLT3 and, in pediatric patients with AML, ITDs are present in 5% to 16% and ALMs are present in 2% to 5% of cases. Several small-molecule inhibitors of the FLT3 tyrosine kinase have been developed, some of which are effective against only the ITDs and some of which are effective against both the ITDs and the ALMs. These inhibitors are now in various phases of clinical trials. Overall, they appear to be well tolerated and to induce a decrease in blasts, but as single agents they generally do not result in durable remissions. The COG completed a pilot study of lestaurtinib in relapsed AML. The drug was well tolerated, but inadequate FLT3 inhibitory plasma concentrations were achieved and the study was closed prior to dose escalation, owing to concerns regarding drug availability. Quizartinib is a tyrosine kinase inhibitor that has shown promise in relapsed adult AML with FLT3 -ITD with clearance of marrow blasts but incomplete count recovery. Rapid resistance to quizartinib is an emerging problem but suggests that the short-term benefits observed are bona fide therapeutic responses. A phase I study conducted by the Therapeutic Advances in Childhood Leukemia and Lymphoma (TACL) consortium demonstrated the tolerability of quizartinib together with cytarabine and etoposide in relapsed and refractory AML and ALL, with remissions both with and without count recovery in four patients, including three with AML and FLT3 -ITD. Sorafenib is another tyrosine kinase inhibitor that has had documented activity as both a single agent and with cytotoxic therapy in relapsed AML, although several of the mutations that are resistant to quizartinib are also resistant to sorafenib. Sorafenib is currently being incorporated upfront for de novo AML patients with FLT3 -ITD and high allelic ratio on COG study AAML1031 and SJCRH AML08. With the use of these agents as frontline therapy, it is uncertain whether they will play a significant role in relapsed disease. However, in cases in which a patient without a prior FLT3 -ITD at diagnosis develops such a lesion at relapse, FLT3 inhibitors should be considered as part of the reinduction-remission strategy. Finally, ponatinib is another new tyrosine kinase inhibitor with activity against FLT3 -ITD, including those with mutations resistant to other inhibitors, and an impressive response rate in refractory CML harboring resistant ABL mutations. This agent may have more broad potential in pediatric myeloid disease; however, the drug has recently been issued a Black Box warning by the FDA due to an association with life threatening thromboses.
Farnesyltransferase Inhibitors.
Mutations in RAS itself, as well as perturbations in the RAS pathway, are associated with AML. The RAS protein requires isoprenylation by farnesyltransferase to bind to the plasma membrane properly and be fully active. Several farnesyltransferase inhibitors have been developed with the goal of inhibiting upregulated or dysregulated RAS activity. Because these drugs do not specifically target a known mutation in RAS and have effect on other proteins that also require farnesylation, they can be effective in patients with or without RAS mutations. Tipifarnib (R115777, Zarnestra) as an oral agent has shown activity in adult patients with refractory AML and in older adult patients with de novo AML, with a few transient complete responses, even when used as a single agent. The drug was generally well tolerated with toxicities of myelosuppression, neurotoxicity, and reversible renal toxicity. In pediatrics, a phase I trial of tipifarnib for refractory solid tumors and neurofibromatosis type 1–related plexiform neurofibromas and a phase II trial for brain tumors both have shown that the drug was tolerated well overall, with dose-limiting toxicities of myelosuppression, rash, nausea, and vomiting; one patient also developed a seizure. A phase I trial in patients with refractory leukemia was completed by COG in 2005. The drug has also been used safely in combination with capecitabine for children with advanced solid tumors. Currently, trials are ongoing combining tipifarnib with chemotherapy for adults with AML as well as a COG trial combining tipifarnib with cis -retinoic acid, cytarabine, and fludarabine with HSCT for children with JMML. Two other farnesyltransferase inhibitors, lonafarnib (Sarasar, SCH66336) and BMS-214662 have also shown some promise in adults with myeloid disorders, but there are as yet no data for pediatric AML.
Histone Deacetylase Inhibitors.
Double-stranded DNA binds to histones to form compact nucleosomes when the DNA is not being actively transcribed. Acetylation of lysine residues by histone acetyltransferases allows the release of DNA from histones for transcription. Deacetylation by histone deacetylase (HDAC) reverses the process. Inappropriate recruitment of HDACs leading to gene silencing has been demonstrated in the CBF leukemic cells bearing translocations t(8;21) and inv(16), as well as in APL cells bearing t(15;17). The use of HDAC inhibitors may allow for renewed expression of silenced genes that when activated can induce differentiation or cell death. Several HDAC inhibitors have been tested in clinical trials in AML patients, including the antiseizure medication valproic acid. It has been used in clinical trials for adults with MDS or AML as monotherapy and in combination with ATRA, 5-aza-2′-deoxycytidine (DAC), as well as 5-azacytidine and ATRA together. It was generally well tolerated, with toxicities of myelosuppression, neurotoxicity, and sedation. Some transient responses were seen, even when valproic acid was used as monotherapy, but it was most effective when used in combination with ATRA and 5-azacytidine. A more potent HDAC, depsipeptide, has been used in clinical trials and has had more toxicity, including fatigue, nausea, and constitutional symptoms. In these trials, it has shown some promise in T-cell lymphoma but thus far only modest antileukemia activity. Vorinostat (suberoylanilide hydroxamic acid [SAHA]) is an HDAC inhibitor that has been under discussion for incorporation in pediatric relapse or refractory AML studies. Romidepsin is a newer HDAC inhibitor also under consideration (T. Cooper, personal communication, May 2013). HDAC inhibitors and DNA methyltransferase inhibitors as combination therapy are also being evaluated. In some trials this seems to be a safe and effective strategy, but in at least one trial using decitabine and valproic acid the combination did not show an improved response rate versus decitabine alone and proved too toxic because of encephalopathy. However, this approach may prove more fruitful in the combination of novel HDAC inhibitors and DNA methyltransferase inhibitors with less overlapping toxicities and in subsets of AML patients found to have particularly high levels of methylation and activity of methyl transferase enzymes. In particular, these therapeutic strategies may be specifically applicable for adult patients harboring DNMT3A and TET2 mutations and MLL rearrangements in pediatric AML.
Apoptosis Inhibitors.
BCL2 is an inhibitor of apoptosis that is highly expressed in some patients with AML and is associated with a poor prognosis. A BCL2 antisense oligonucleotide has been tested as a single agent and shown to induce transient remission in some AML patients. It has also been shown to be safe and effective in combination with cytarabine and daunorubicin. This agent is now being used in clinical trials of AML through the Cancer and Leukemia Group B (CALGB) as part of combination therapy during induction and consolidation. Specifically, a trial combining the pan-BCL2 inhibitor obatoclax with vincristine, doxorubicin, and dexrazoxane for relapsed solid tumors and leukemias has just been completed by the COG and results are pending.
Other Targeted Therapies.
A number of new promising agents are emerging and being considered for introduction into relapsed AML trials. Some of these agents target recently emerging important molecular pathways in AML cells such as MK2206, which targets the PI3K-AKT pathway, or MK8776, which is a CHK1 inhibitor. AKT pathway activation may contribute to resistance to GMTZ and may be particularly active in AML associated with Down syndrome (see later). Phase I studies of MK2206 have been completed in adults, and a pediatric trial has just been completed. CHK1, or checkpoint kinase 1, regulates the G2 to M phase transition of the cell cycle and may be activated in the phase of DNA damage in AML cells. CHK1-inhibiting agents have been evaluated in a number of adult phase I solid tumor studies and may have efficacy in AML, particularly in children harboring complex karyotypes associated with the highest levels of DNA damage and CHK1 activity. CHK1 has been shown to suppress apoptosis mediated by caspase 2 in the context of DNA damage, and inhibition of CHK1 may restore apoptosis in AMLs with TP53 mutations. Another strategy is to interfere with the interaction of AML cells and their microenvironment through the use of drugs such as plerixafor. Plerixafor binds to and antagonizes the interaction of CXCL12 (also known as SDF1) and its receptor CXCR4 found on HSCs and potentially on LICs, affecting LIC mobilization from the stem cell niche and making these cells more accessible to cytotoxic chemotherapies. Thus this agent is believed to target the AML LIC and make these resistant cells more susceptible to conventional therapies. A recent phase I/II adult trial gave plerixafor prior to cytarabine, etoposide, and mitoxantrone for patients with relapsed AML and found that this combination was well tolerated and resulted in approximately 50% complete remission rates.
Acute Promyelocytic Leukemia
Pathobiology
APL or M3 AML is a subtype of AML defined by a specific morphology, a balanced translocation involving the RARA gene, and a characteristic coagulopathy. Although most patients with APL have the classic t(15;17) resulting in the PML-RARA fusion gene, up to 8% do not. Approximately 5% of patients have a PML-RARA because of an insertion or other variant, 1% of patients have a t(11;17) resulting in a PLZF-RARA, another 1% have a t(5;17) resulting in an NPM-RARA or a t(11;17) resulting in a NuMA-RARA or a STAT5b-RARA , and 1% of patients do not have an identifiable RARA rearrangement. APL provides a clear example of the classic two-hit model for AML (see earlier, “ Molecular Genetics ”). The fusion protein resulting from the pathognomonic translocation in APL, the PML-RARA protein, is an example of a class II mutation, resulting in a block in differentiation. As detailed earlier (see “ Biology ”), this protein has been shown to interfere with normal myeloid cell development through dominant inhibitory mechanisms as a potent transcriptional repressor that recruits a number of other corepressors to cause differentiation arrest at the promyelocyte stage. Mutations in the receptor tyrosine kinase FLT3 occur in up to 43% of cases of APL, and these mutations are classic examples of class I mutations, conferring a proliferative advantage. At diagnosis, APL patients with FLT3 mutations more often present with hyperleukocytosis, microgranular variant morphology, and the bcr3 PML breakpoint. Although FLT3 -ITDs clearly confer a worse prognosis in other subtypes of AML, their role in prognosis for APL is less clear. Some groups have reported a worse prognosis for APL patients with an FLT3 -ITD, whereas other groups have found no statistically significant impact on the generally favorable risk outcome of this disease. A recent report from the COG found FLT3 -ITD or ALM mutations in 40% of children treated on the CALGB 9710 study and identified a strong correlation with higher WBC count and induction death rates in the FLT3 mutant subgroup.
Clinical Presentation and Risk Factors
APL is more common in girls and obese children, and obesity is associated with a higher rate of relapse and differentiation syndrome (see later). Compared with adults, children with APL have a higher incidence of hyperleukocytosis at presentation, microgranular variant morphology, and PML-RARA isoforms bcr2 and bcr3. Moreover, a recent retrospective review of two European studies demonstrated that children younger than age 4 years had a higher incidence of disease relapse than those age 5 to 12, whereas, by contrast, adolescents had higher OS than adults. A presenting WBC count higher than 10,000/µL has been repeatedly shown to carry a worse prognosis, as has a platelet count less than 40,000/µL, leading to the intensification of therapy with additional anthracycline, cytarabine, and/or ATRA for adult patients treated on recent French and Italian APL trials. Based on improved outcomes, on the recently completed COG trial AAML0631, patients with a WBC count greater than 10,000/µL were designated as high-risk patients who were relegated to additional consolidation cycle with high-dose cytarabine. Before the 1970s, bleeding complications caused significant mortality for patients with APL. Measures that have been used successfully to manage the coagulopathy include platelet and cryoprecipitate transfusions, use of low-dose heparin, and prompt initiation of anthracycline-based chemotherapy. However, the most dramatic decrease in early and induction-related deaths has been the result of the addition of ATRA to the induction regimen, which rapidly leads to the resolution of DIC, and therefore has markedly reduced early mortality.
Treatment
All- Trans -Retinoic Acid
With the introduction of ATRA, the treatment and outcome of APL have changed dramatically. Although previously considered primarily a differentiation agent, a number of recent studies in PML-RARA expressing mice and cell lines containing the alternative PLZF-RARA fusion have uncoupled differentiation and leukemic cell death, suggesting that exclusive acceptance of a differentiation paradigm belies ATRA’s true mechanism of action. In keeping with these recent findings, while even low-dose ATRA induces differentiation in APL blasts in vivo and in vitro and induces remission in up to 90% of patients with newly diagnosed APL, these affects are transient without additional chemotherapy or As 2 O 3 (see later). Although ATRA therapy results in a lifting of the repression complex that inhibits APL blast differentiation, higher doses, As 2 O 3 , or chemotherapy is needed to degrade the PML-RARA fusion protein and halt the self-renewal process, ultimately eliminating the malignant clone. Elimination of PML-RARA may further result in more durable cures by targeting the LIC population in APL in which the initial t(15;17) occurred, as well as restoring the normal function of the intracellular PML bodies disrupted by the presence of the fusion protein. Thus recent strategies combine ATRA with conventional chemotherapy, resulting in improved prognosis for patients with APL compared with chemotherapy alone. A long-term analysis has demonstrated significantly higher actuarial EFS rates, lower relapse rates, and improved OS in the group treated with both ATRA and chemotherapy. The addition of ATRA to the chemotherapy regimen has been particularly effective for pediatric patients. The APL93 trial used ATRA in combination with daunorubicin and cytarabine in induction, and the children in this trial had a complete remission rate of 97%. The PETHEMA and GIMEMA-AIEOPAIDA groups have completed pediatric trials using ATRA and idarubicin as induction chemotherapy, with reported CR rates of 92% to 96%. As such, this lower dose has become the standard for current North American APL studies in children. Current recommendations are to initiate ATRA therapy early, at the time when a diagnosis of APL first is suspected, to correct and prevent devastating consequences from an evolving coagulopathy.
One potentially life-threatening complication of ATRA therapy is the retinoic acid syndrome. This syndrome occurs in approximately 25% of patients and is characterized by fever, respiratory distress, pleural or pericardial effusions, edema, hypotension, and, in some cases, renal failure. In most patients, the symptoms are preceded by an increasing WBC count. Retinoic acid syndrome can be life threatening, and the mortality rate ranges from 8% to 15% in different reports. The syndrome is also associated with a higher risk for bone marrow and extramedullary relapse. The use of chemotherapy concurrently with ATRA therapy during induction, particularly in patients with high presenting WBC counts, results in a significantly lower incidence of fatal retinoic acid syndrome ; and given the dose dependence, ATRA syndrome now occurs less frequently with the use of 25 mg/m 2 as the standard therapeutic dosing (see later). The use of high-dose corticosteroids at the first sign of symptoms has also been shown to be effective in preventing ATRA syndrome and reducing its mortality rate. Another serious complication of ATRA therapy is pseudotumor cerebri, presenting as headache and papilledema. True pseudotumor cerebri is observed in up to 16% of children, and an additional 39% of children develop headaches without other signs of increased intracranial pressure. Pseudotumor cerebri may require treatment with diuretics such as acetazolamide or serial lumbar punctures to reduce intracranial pressure; and, in some cases, discontinuation or dose reduction of ATRA is required. Close follow-up with an ophthalmologist is advised because these patients may suffer visual loss, which in rare cases may be irreversible. Because of the high rate of ATRA-related side effects, the AML-BFM study group studied dose reduction of ATRA during induction from 45 to 25 mg/m 2 /day. They found that this dose was still effective, although in pediatric patients there was still a very high rate of CNS side effects, with 57% of children exhibiting headache, increased intracranial pressure, or frank pseudotumor cerebri. The CALGB trial C9710 included pediatric patients and used the higher dose of ATRA, but recent data demonstrated no additional benefit and only increased toxicity compared with 25 mg/m 2 dosing. Therefore this lower dose was incorporated into the recently closed first exclusively pediatric COG APL study, AAML0631, and this dose is now considered the standard of care in pediatric APL.
Arsenic Trioxide
Arsenic trioxide (As 2 O 3 ,) has been used for decades in China as treatment for leukemia. The precise mechanism of action is actively being studied, but As 2 O 3 appears to induce both differentiation and apoptosis of APL blasts and to degrade the PML-RARA fusion protein while not affecting transcriptional regulation per se. As 2 O 3 appears to induce nonterminal differentiation followed by apoptosis via the caspase pathway. When As 2 O 3 was used as a single agent in patients with APL, remission rates of 65% to 85% were reported and the 10-year survival was as high as 30%. Later studies done in China and the United States showed favorable results, with moderate toxicity, using As 2 O 3 in patients with relapsed APL who had previously received ATRA therapy. Several studies have shown promising results using As 2 O 3 in remission induction and consolidation therapy for patients with newly diagnosed APL. One small series of 11 children with newly diagnosed APL treated with As 2 O 3 as a single agent showed mild toxicity, including leukocytosis, skin changes, and mild neuropathy. CR was induced in 91% of these patients, with 1 patient dying of cerebral hemorrhage during induction. With a relatively short median follow-up of 30 months, the DFS was 81% with only 1 relapse; the patient who experienced relapsed achieved a second CR, and thus the OS rate remains 91%. On the U.S. phase I pediatric study of As 2 O 3 for relapsed leukemia, 13 patients with relapsed APL received As 2 O 3 intravenously daily at 0.15 mg/kg/dose for 5 days per week for 4 weeks with a 2-week break with maximum of 70 doses. A CR rate of 85% was observed after a median of 20 doses. Dose-limiting toxicities included rare vascular leak, anorexia, neuropathic pain, and QTc prolongation (see later).
ATRA and As 2 O 3 have been used safely together as induction therapy in children and adults. The drugs may have a synergistic effect when used together; one study showed a shorter median time to CR in the group receiving the combination (40.5 days for ATRA alone, 31 days for As 2 O 3 alone, 25.5 days for ATRA with As 2 O 3 ). The combination also provided longer duration of remission in this study. Because of these promising data, several groups have included As 2 O 3 as part of induction or consolidation therapy for newly diagnosed pediatric patients with APL, with the hope of reducing the total cumulative dose of anthracycline exposure. As 2 O 3 was used in a randomized fashion during consolidation in the CALGB 9710 trial that included pediatric patients through the COG and demonstrated a dramatic improvement in outcome. Patients 15 years of age and older who had the addition of As 2 O 3 after a common ATRA and chemotherapy induction demonstrated significantly improved 3-year EFS (77% vs. 59%) and OS (86% vs. 77%) compared with patients who received ATRA and chemotherapy alone. These results led to the nonrandom incorporation of As 2 O 3 into the successor COG study AAML0631, such that all patients nonrandomly received As 2 O 3 in a dose of 0.15 mg/m 2 for 5 days a week for five courses during consolidation therapy.
As 2 O 3 therapy is generally well tolerated, often with only minimal and reversible toxicity. However, care must be taken with its use, because it can produce a prolonged QTc interval, which may be asymptomatic but can progress to torsades de pointes and fatal cardiac arrhythmia. In one retrospective analysis, the rate of arrhythmia was significantly higher in African American patients (3 of 4) versus non–African-American patients (1 of 73). QTc intervals should be followed closely by weekly electrocardiography during As 2 O 3 treatment, and hypokalemia and hypomagnesemia should be meticulously avoided. Because of the induction of cellular differentiation, As 2 O 3 commonly results in a condition similar to retinoic acid syndrome, most often characterized by fluid retention and pleural and pericardial effusions. This may be successfully treated in the same way as ATRA-induced retinoic acid syndrome. As 2 O 3 can also cause a significant polyneuropathy, particularly when given in repeated courses. Less dangerous side effects include skin changes with rash, hyperpigmentation, keratosis, and transient liver function test abnormalities. In a COG phase I study of As 2 O 3 in children with refractory or relapsed acute leukemia, dose-limiting toxicities included prolonged QTc, pneumonitis, and neuropathic pain, whereas non–dose-limiting toxicities included elevated liver function test results, nausea, vomiting, abdominal pain, constipation, electrolyte changes, hyperglycemia, dermatitis, and headache.
Owing to their synergistic effects, ATRA and As 2 O 3 were both used upfront on the AAML0631 study and were generally well tolerated. Although used together with chemotherapy on this recently completed trial, the efficacy of this combination and recent adult studies demonstrating the ability of this combination therapy to lead to sustained remission without additional cytotoxic chemotherapy are likely to lead to future trials in which additional chemotherapy is significantly reduced or even eliminated to attain a first CR at least in standard patients. Most striking was the recent APL0406 study, which randomized adult APL patients to ATRA and As 2 O 3 or ATRA and idarubicin in induction and found a 2-year EFS of 97% versus 86.7% in favor of the ATRA/As 2 O 3 trial arm. Thus the addition of anthracyclines and cytarabine may be reserved for APL patients designated high risk at diagnosis based on WBC count, those with more resistant disease (PML-RARA positive post-consolidation [see later]), or first recurrence.
Cytarabine
Although cytarabine is the backbone of induction chemotherapy for other subtypes of AML, its role in APL has been less clear. The PETHEMA group completed a pediatric trial using ATRA and idarubicin as induction chemotherapy without cytarabine, and the CR rate was 92%. The GIMEMA-AIEOPAIDA protocol also used ATRA and idarubicin during induction without cytarabine and showed a 96% CR rate in children. However, this question was subsequently studied in a randomized clinical trial in which patients younger than 60 years and with a presenting WBC count less than 10,000/µL were randomly assigned in induction to ATRA and daunorubicin, with or without cytarabine. CR rates were comparably high for the two groups—99% with cytarabine and 94% without; however, in the cytarabine arm, the relapse rate was significantly lower (4.7% vs. 15.9%; P = .011), the EFS higher (93.3% vs. 77.2%; P =.0021), and the OS higher (97.9% vs. 89.6%; P = .0066). Of note, the current trials all use high cumulative anthracycline doses to achieve excellent OS rates but there is concern about long-term cardiac toxicity, particularly in pediatric patients. Considering the improved results with the addition of cytarabine to the induction and consolidation regimens, several groups have been studying the use of more cytarabine with less anthracycline in pediatric patients with APL. On AAML0631, 6 g/m 2 of cytarabine was given during the second consolidation cycle for all patients, with an additional 6 g/m 2 of cytarabine for high-risk patients. The cumulative anthracycline dose was decreased from 655 mg/m 2 on prior trials to 355 mg/m 2 for standard-risk patients and 405 mg/m 2 for high-risk patients.
Maintenance Therapy
Another difference between APL and other subtypes of AML historically has been an identified benefit from maintenance therapy. The APL93 trial randomized pediatric patients after consolidation to an arm with no further therapy or to arms with 2 years of intermittent ATRA, continuous chemotherapy with 6-mercaptopurine and methotrexate, or both ATRA and chemotherapy. Although 23% of pediatric patients in this trial experienced relapse, none of the patients who received both chemotherapy and ATRA during maintenance therapy did so. In this trial, all patients with relapsed APL achieved a second CR. The 5-year EFS was 71%, and OS was 90%. The PETHEMA group also used ATRA plus 6-mercaptopurine and methotrexate during 2 years of maintenance therapy for pediatric patients and demonstrated a DFS of 82% and an OS of 87%. Since these earlier studies, all recent trials for APL have included a maintenance phase of therapy. To be congruent with parallel European studies, the recently completed AAML0631 study included 2 years of maintenance therapy with ATRA, 6-mercaptopurine, and methotrexate, whereas the CALGB study only included a single year of maintenance with the same agents. However, the future of maintenance therapy in APL is uncertain. The need for chemotherapy versus ATRA alone in maintenance therapy has been raised. Preliminary results of upfront ATRA and As 2 O 3 in both Italian and M.D. Anderson Cancer Center adult trials without inclusion of maintenance therapy have prompted consideration of elimination of maintenance therapy altogether. Inclusion of maintenance therapy and whether any chemotherapy is necessary will be key therapeutic goals explored in upcoming trials.
Targeted Therapy
With the use of ATRA and As 2 O 3 , targeted therapy for APL has become a reality. Other targeted agents are also being used successfully in APL. GMTZ (see earlier) is a recombinant, humanized, anti-CD33 monoclonal antibody linked to the tumor antibiotic calicheamicin. APL cells express very high levels of CD33, making them an attractive target for GMTZ. GMTZ has been used successfully as a single agent to induce remission in patients with multiply relapsed APL. In a small series of 12 patients, GMTZ was used safely in combination with ATRA with or without idarubicin, depending on the extent of MRD, as induction therapy for patients with de novo APL with a CR rate of 84%. Some groups are now studying the use of GMTZ as induction therapy in larger numbers of patients. Although not currently used initially for children with APL, GMTZ may be included for the management of high-risk patients and standard-risk patients who have a rise in their WBC count over 10,000/µL during induction therapy or, alternatively, as a strategy to induce remission after relapse in patients who have already received ATRA, As 2 O 3 , and cytotoxic chemotherapy.
As discussed earlier, mutations in FLT3 have been identified in up to 43% of patients with APL, making the use of FLT3 inhibitors an attractive option. Most FLT3 inhibitors are oral agents with little toxicity. In one mouse model of APL with the FLT3 mutation, treatment with doxorubicin alone had no impact on survival, but treatment with the FLT3 inhibitor SU11657, with or without doxorubicin, resulted in prolonged survival. In another mouse model, treatment with ATRA and SU11657 resulted in the rapid regression of APL. There is considerable interest in using FLT3 inhibitors in combination with ATRA and other targeted agents to improve therapy for patients with APL.
Hematopoietic Stem Cell Transplantation
Because the combination of chemotherapy and ATRA, and now ATRA and As 2 O 3 , results in such high cure rates for APL, HSCT is not recommended for patients in first CR, even with a matched sibling donor. There is evidence for prolonged remission without HSCT, even after relapse for patients treated with agents such as ATRA, As 2 O 3 , and GMTZ, especially when combined with chemotherapy. Both autologous and allogeneic HSCT have been used successfully in patients with relapsed APL. For autologous HSCT to be successful, peripheral blood stem cells or bone marrow cells must be harvested once the patient has achieved negative MRD by PCR assay. The GVL effect appears to play a role in the allogeneic HSCT treatment of APL, as evidenced by the fact that relapse rates are significantly lower after allogeneic HSCT compared with autologous HSCT and that patients who suffer a molecular relapse after allogeneic HSCT may achieve remission after the withdrawal of immunosuppression. However, despite the lower relapse rate, because of significantly higher transplantation-related mortality (TRM) after allogeneic HSCT, OS in adults is higher after autologous HSCT than after allogeneic HSCT (postallogeneic HSCT: DFS, 92.3%; EFS, 52.2%; OS, 51.8%; TRM, 39%; and postautologous HSCT with negative preharvest PCR: DFS, 87.3%; EFS, 76.5%; OS, 75.3%; TRM, 6%). In pediatric patients, allogeneic HSCT is much better tolerated. A retrospective analysis demonstrated no difference in OS after allogeneic HSCT versus autologous HSCT, although the allogeneic HSCT group had a lower relapse rate and higher TRM (postallogeneic HSCT: RR, 10%; EFS, 71%; OS, 76%; TRM, 19%; and postautologous HSCT: RR, 27%; EFS, 73%; OS, 82%; TRM, 0%). Given these data, in adults, autologous HSCT is usually the preferred choice versus allogeneic HSCT for patients in second CR, whereas in children, allogeneic HSCT is often the preferred choice, particularly if a fully matched sibling donor is available.
Central Nervous System Involvement
In patients with APL, CNS disease at the time of diagnosis is rare and, because of coagulopathy, diagnostic lumbar puncture prior to the initiation of therapy is not safe for most patients. Therefore if APL is suspected, lumbar punctures should not be performed until the coagulopathy is adequately corrected and ATRA initiated. In the pre-ATRA era, CNS relapse was rarely observed in APL patients. However, while the survival rate for APL improved dramatically with the introduction of ATRA, an increasing number of CNS relapses have been reported. Unlike other subtypes of AML, most APL regimens do not include CNS prophylaxis. CNS relapse is more common in patients with a presenting WBC count higher than 10,000/µL, with microgranular variant morphology, and with bcr3 PML-RARA isoform, all of which are also associated with the presence of an FLT3 ITD and all of which are more common in pediatric patients. Although there was initially concern that ATRA itself was contributing to CNS relapses, it now seems that ATRA does not penetrate the CNS well and, with more patients surviving APL therapy, this likely contributes to the higher rate of CNS relapse currently observed. Several groups have now initiated a screening lumbar puncture at the time of CR and use prophylactic intrathecal chemotherapy, usually with cytarabine and methotrexate, for patients at high risk for CNS relapse. On AAML0631, prophylactic cytarabine was given intrathecally in consolidation 2, 3, 4 (for high-risk patients) and the first maintenance course but at a lower dose of 30 mg (25 mg for children >2 years of age but <3 years of age) compared with the 70-mg given as standard dosing in other subtypes of AML. Given the infrequency of CNS positivity in APL, future studies may eliminate intrathecal therapy entirely.
Minimal Residual Disease Monitoring
Detection of the PML-RARA fusion gene by reverse-transcriptase PCR (RT-PCR) has become a valuable tool for measuring MRD in APL. RT-PCR positivity at the end of induction does not seem to portend a higher risk for relapse and therefore should not be checked at that time point, but RT-PCR positivity after consolidation does carry a significantly higher risk for relapse. RT-PCR positivity during maintenance or after therapy is indicative of impending relapse in almost all patients. MRD analysis is being used to identify high-risk groups that require more intensive therapy for cure. When detected early by RT-PCR, molecular relapse may also be easier to treat than frank morphologic relapse because of the lower disease burden and the treatment of a healthier asymptomatic patient. In many studies, RT-PCR analysis is carried out on bone marrow cells and evidence regarding analysis of peripheral blood is not as clear, thus PML-RARA surveillance on AAML0631 mandated bone marrow samples. However, the frequency of relapse after therapy including As 2 O 3 is so rare for standard-risk patients that whether follow-up on future trials will require the invasiveness of bone marrow evaluations remains to be determined. There is also controversy regarding the threshold for a “positive” result; some groups do not consider a result a true positive unless there are two positive results separated by 2 to 4 weeks. However, in the event that suspicion is significant for recurrence based on a rising level of PML-RARA transcript in the peripheral blood, a bone marrow evaluation should be conducted to look for molecular, cytogenetic, and hematologic signs of relapse.
Relapsed Disease
In the current ATRA and As 2 O 3 era, relapsed APL is a rare occurrence and there is limited experience on what therapeutic strategy to undertake in children who have already received ATRA, As 2 O 3 , and chemotherapy. Given their efficacy, reinduction with combination ATRA and As 2 O 3 is a reasonable strategy if previously well tolerated, although it is unclear whether the addition of chemotherapy provides any further benefit or merely contributes to toxicity. Currently, patients with relapsed APL may have already seen a significant cumulative dose of anthracycline, such that additional anthracycline may push the normally accepted lifetime ceilings with which most clinicians are comfortable. However, the concomitant use of cardioprotectants, such as dexrazoxane, may provide the opportunity to provide further anthracycline therapy at relapse. Furthermore, if future studies show the initial use of chemotherapy and anthracyclines in particular may be significantly reduced owing to the efficacy of combination ATRA and As 2 O 3 , it may enable chemotherapy to be reserved for relapses and refractory cases. Regardless, particularly with early relapse, given the opportunity for GVL effect and the improvement in both high-resolution HLA typing and better supportive care, after reinduction, best available donor allogeneic transplant would be recommended by most experts. However, on prior studies, TRM with allogeneic HSCT was higher than with autologous transplant, although there was no significant difference in 5-year OS or EFS, so this decision should not be made lightly. With later relapses, the ability to induce a second sustained CR with ATRA, As 2 O 3 , and chemotherapy without HSCT may be a feasible alternative, although with small numbers of patients most of these data are anecdotal.
Down Syndrome with Transient Disorder and Acute Myelogenous Leukemia
Down Syndrome with Transient Myeloproliferative Disorder
Pathobiology
Down syndrome, characterized by trisomy 21, is the most common congenital chromosomal abnormality, occurring in an estimated 1 in 600 to 1000 live births. Up to 10% of infants with Down syndrome are born with a transient myeloproliferative disorder (TMD) (also referred to as transient leukemia [TL] or transient abnormal myelopoiesis [TAM]). TMD is characterized by the presence of immature megakaryoblasts in the liver, bone marrow, and peripheral blood. The disorder usually spontaneously regresses within 3 months. By morphology, TMD blasts are identical to AMKL blasts and, even though in most cases they regress, up to 30% of Down syndrome infants with TMD subsequently develop AMKL within 3 years. Studies have shown that TMD blasts are clonal and that the same clone subsequently evolves into AMKL in a subset of patients.
In 2002, Wechsler and colleagues first reported mutations in exon 2 of GATA1 in Down syndrome–associated AMKL. Soon thereafter, several groups reported that GATA1 mutations were detectable in almost all patients with Down syndrome–associated TMD. Interestingly, GATA1 mutations have also been detected in approximately 10% of Down syndrome patients who have never been diagnosed with a hematologic abnormality. It is not clear whether these patients have a history of subclinical TMD that regressed without detection or if the mutation simply did not result in the TMD phenotype in these cases. Normal GATA1 plays a vital role in the maturation of erythroid cells and megakaryocytes. The GATA1 mutations in Down syndrome TMD and AMKL result in GATA1s, a truncated protein lacking the amino-terminal activation domain. GATA1s appears to be a normal isoform of GATA1 and has been detected in human hematopoietic cell lines and mouse fetal liver. However, because GATA1 is an X-linked gene, only the truncated GATA1s is expressed in Down syndrome TMD and AMKL blasts. It has been hypothesized that the presence of a trisomy 21 in conjunction with a GATA1s allows abnormal proliferation of megakaryoblasts, but only in the fetal hematopoietic system (i.e., the fetal liver); thus TMD resolves with the loss of fetal hematopoiesis. Although the combination of trisomy 21 and a GATA1 mutation appears sufficient to result in TMD, this is not sufficient to result in frank leukemia, and only if additional mutations are accumulated over time will the disease progress to frank AMKL.
Wilms tumor gene (WT1) levels detected by PCR assay are elevated in patients with TMD. Levels normalized in four of five patients studied who had regression of TMD, with no further sequelae, but did not normalize in the fifth, who went on to develop AMKL at 11 months of age, making this an attractive marker for MRD and prognosis. Both gain- and loss-of-function mutations in Janus kinase 3 (JAK3) have been identified in a subset of patients with TMD, although the prognostic relevance of this is not yet known.
Clinical Presentation
Because Down syndrome infants with TMD are often asymptomatic, the incidence of 10% may be an underestimate, because many patients may have TMD that spontaneously regresses, without ever being detected. For this reason, many physicians obtain a routine screening complete blood cell (CBC) count at birth for all infants with known or suspected Down syndrome. However, some infants with Down syndrome have a dramatic presentation of TMD at or shortly after birth, with hydrops fetalis, pleural and pericardial effusions, ascites, and massive hepatosplenomegaly. These patients may rapidly progress to liver fibrosis or multisystem organ failure, both of which are most often fatal. In the Pediatric Oncology Group (POG) trial 9481, 48 children with TMD were followed prospectively; 19% developed life-threatening disease with hepatic fibrosis or cardiopulmonary failure, with an overall mortality at 3 months of age of 10.4%. Another retrospective analysis from Japan has shown that 22.9% of patients with TMD die before the age of 6 months, and the main causes of death were hepatic or cardiopulmonary failure. In this study, early gestational age, WBC count greater than 100,000/µL, high percentage of peripheral blasts, elevated aspartate aminotransferase (AST), elevated direct bilirubin, and low Apgar score were significantly associated with poor survival. They noted that only 7.7% of term Down syndrome infants with a WBC count less than 100,000/µL died, whereas 54.5% of preterm infants with a WBC count higher than 100,000/µL died, suggesting that risk stratification may be possible to determine high-risk subgroups of patients who require earlier and more aggressive therapy.
Treatment
Because most patients with TMD will exhibit spontaneous regression, treatment often consists of supportive care only. Many groups, including COG protocols 2971 and 08B1, have recommended supportive care only for TMD patients unless they exhibit signs or symptoms of life-threatening disease, including hyperviscosity, with a total blast count higher than 100,000/µL, organomegaly causing respiratory compromise, congestive heart failure not caused by a congenital heart defect, hydrops fetalis, life-threatening hepatic dysfunction, defined as significant (grade 4 level toxicity) DIC, hyperbilirubinemia, ascites, or transaminitis. For patients who require therapy, the choices include leukapheresis, exchange transfusion, or low-dose cytarabine. Low-dose cytarabine has been effective when used early in critically ill patients. In patients with hyperleukocytosis or who require therapy with cytarabine, treatment to prevent the consequences of rapid tumor lysis (see earlier) should be promptly initiated.
Down Syndrome with Acute Myelogenous Leukemia
Pathobiology
Children with Down syndrome have a markedly increased risk for leukemia during the first 10 years of life. Acute leukemia is 10 to 20 times more common in children with Down syndrome than in children in the general population, and leukemia in Down syndrome patients younger than 4 years is most commonly AML (specifically AMKL), whereas leukemia in older children with Down syndrome is most commonly ALL. The ratio of ALL to AML is approximately 1 : 1 in children with Down syndrome, but in the first 4 years of life, AML is 100 times more common than ALL. Although AMKL is a rare subtype of AML in the general population, it is the predominant form of AML in children with Down syndrome, making Down syndrome patients 500 times more likely than other children to develop AMKL. Unlike other children with AML, AMKL in children with Down syndrome often manifests after a period of myelodysplasia, usually characterized by months of thrombocytopenia and later by anemia. Most cases of AMKL in Down syndrome patients are preceded by TMD (see earlier). Several studies have confirmed that when there is a proven antecedent TMD, the same TMD clone subsequently evolves into AMKL.
Also characteristic of AMKL in Down syndrome are GATA1 mutations (see earlier). Because GATA1 encodes a transcription factor essential for megakaryocyte development, samples from patients with AMKL were studied for evidence of GATA1 mutations. Mutations in exon 2 of GATA1 were detected in AMKL blasts from Down syndrome patients but not in other subtypes of AML in Down syndrome patients or in AMKL blasts from non–Down syndrome patients. Several subsequent studies have confirmed GATA1 mutations leading to a premature stop codon in almost all cases of Down syndrome–associated AMKL. GATA1 mutations have been identified in the blood spots of three of four Down syndrome patients with AMKL who had not been previously diagnosed with TMD, suggesting that in most cases of AMKL the mutation has been present since birth. Down syndrome patients who develop AML after the age of 4 years do not usually have GATA1- mutated AMKL but have AML more typical of the general pediatric population (non–Down syndrome AML). This disease is not as sensitive to chemotherapy and does not carry the exceptionally good prognosis carried by Down syndrome AMKL (see later).
Additional mutations beyond the GATA1 mutation in the presence of trisomy 21 are necessary to produce AMKL. The good-risk translocations common in other forms of pediatric AML, including t(8;21), t(15;17), inv(16), and t(9;11), are almost never seen in Down syndrome AMKL, although they are more commonly reported in Down syndrome patients who develop AML after the age of 4. Other than trisomy 21, trisomy 8 is the most common cytogenetic abnormality in Down syndrome AMKL. Other translocations commonly reported in AMKL in the absence of Down syndrome, including t(1;22) and t(1;3), are only rarely documented in Down syndrome AMKL. Monosomy 7, a widely accepted marker of pediatric and adult high-risk AML in the absence of Down syndrome, is also seen in 3.5% to 10% of patients with Down syndrome and AML and, although data are limited, it is generally associated with a worse outcome (see later). Approximately 25% of patients have no cytogenetic abnormalities other than the constitutional trisomy 21, although some patients will exhibit tetrasomy 21. As in TMD, overexpression of WT1 and mutations in JAK3 have been reported in Down syndrome AMKL. More recently, next generation sequencing approaches have identified a number of potential somatic mutational drivers that might contribute to the progression of TMD to AMKL including mutations in the cohesin complex; the epigenetic regulator, EZH2; as well as activating lesions in the RAS, WNT, mitogen activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways.
Clinical Presentation
Children with Down syndrome who develop AML present at a younger age than average for childhood AML, with 60% to 70% of patients diagnosed when they are younger than 2 years. Down syndrome patients generally present with lower total WBC counts (median, 7,000 to 10,000/µL) and lower platelet counts (median, 25,000 to 30,000/µL) than other children. Hepatosplenomegaly is more common in Down syndrome patients with AML than in other children, whereas lymphadenopathy and CNS involvement are less common. Down syndrome AMKL is frequently preceded by TMD, which is almost absent in non–Down syndrome patients, or MDS, which is far less common in non–Down syndrome patients. Older Down syndrome patients with AML have presenting signs and symptoms that more closely mirror those of the general population, reflecting the virtual lack of AMKL with GATA1 mutations in patients older than 5 years.
Treatment and Outcome
Many children with Down syndrome have been entered into large clinical trials since 1982, when legislation was passed that prohibits withholding medical intervention from children with disabilities. In the early clinical trials that included patients with Down syndrome and AML, the outcome was poor because of excessive toxicity. The associated congenital abnormalities, increased susceptibility to infection, and altered drug metabolism in patients with Down syndrome made it challenging to find appropriate therapy for this group. However, while general medical care for children with Down syndrome has improved, the outcome for children with AML and Down syndrome has also improved. Multiple studies from around the world have consistently demonstrated that Down syndrome AML patients have a better overall prognosis than children with AML in the general population. A remarkable feature of AML in children with Down syndrome is its extraordinary responsiveness to AML chemotherapy. Compared with non–Down syndrome AML, leukemic blasts are 4.5-fold and 12-fold more sensitive in vitro to cytarabine (median half maximal inhibitory concentration [IC 50 ], 77.5 vs. 350.9 nM) and daunorubicin (median IC 50, 5.8 vs. 71.2 nM), respectively. The increased sensitivity of Down syndrome AML blasts to cytarabine has been explained by a 5.2-fold greater accumulation of the active drug metabolite cytarabine-CTP compared with non–Down syndrome AML blasts. Down syndrome AMKL blasts are significantly more sensitive to other chemotherapy agents, including amsacrine (16-fold), etoposide (20-fold), 6-thioguanine (3-fold), busulfan (5-fold), and vincristine (23-fold). In addition to constitutionally expressed genes encoded on chromosome 21, which modulate drug metabolism in Down syndrome AML blasts, such as cystathionine-β-synthase (CBS), the expression of mutant GATA1 protein itself contributes to increased drug accumulation. In addition, the cytidine deaminase (CDA) levels in Down syndrome AMKL blasts are low, leading also to increased sensitivity to cytarabine. The CDA promoter contains several binding sites for GATA1, suggesting that GATA1 mutations may contribute to the sensitivity of these blasts.
Observations of increased in vitro sensitivity of Down syndrome AMKL blasts to cytotoxic drugs have been complemented by pioneering studies in which very low doses of cytarabine, which were administered over a prolonged period of time (10 mg/m 2 subcutaneously for 12 hours for 7 days every 2 weeks for 2 years), resulted in durable remissions in as many as 67% of cases (intention to treat analysis ). Although outcomes of the very-low-dose cytarabine regimen eventually did not match those of standard dose cytarabine, the observation highlights a unique degree of drug sensitivity of Down syndrome AMKL blasts to cytarabine that is not encountered in pediatric non–Down syndrome AML. Recently, Japanese investigators showed that high-dose cytarabine could successfully be eliminated from a treatment protocol for Down syndrome AML while maintaining a 4-year EFS of 83%. In parallel, studies maximizing dose intensity for the treatment of AML, although successful in the general pediatric population, did not translate into any improvements in EFS for patients with Down syndrome and AML owing to excessive mortality during induction therapy in this group. In the German collaborative AML-BFM93 protocol, children with Down syndrome fared worse than other patients because of an increased frequency of infectious complications, but none of the patients experienced relapse and EFS was approximately 70%. Therefore, in the subsequent AML-BFM98 protocol, Down syndrome patients received a reduced intensity regimen and the EFS improved dramatically, to almost 90%. In the Nordic Society for Paediatric Haematology and Oncology-Acute Myeloid Leukaemia (NOPHO-AML) trials, the earlier, more intensive NOPHO-AML88 trial showed a high death rate from infectious complications for Down syndrome patients with AML, with an EFS of only 47%, compared with much better outcomes in the later, less-intensive NOPHO-AML93 trial, with an EFS of 85% (76% for those who received full-dose therapy and 92% for those who received reduced-dose therapy). The MRC trials showed similar results with Down syndrome patients receiving the same standard therapy as other patients, resulting in a higher rate of death from toxicity but lower relapse rate when compared with other children. Perhaps the most striking example comes from the CCG 2891 study. In this study, all children were randomly assigned to receive induction chemotherapy with intensive timing (i.e., proceeding with the second cycle, regardless of counts) versus standard timing (i.e., awaiting count recovery before initiating the second cycle). The Down syndrome patients in the intensive timing arm of the study suffered a 32% mortality rate compared with an 11% mortality rate for the remainder of the patients, prompting early closure of that study arm for Down syndrome patients. Standard timing induction for Down syndrome patients resulted in an impressively high CR rate of 95% (2.4% with toxic death and 2.4% with resistant disease). Postremission consolidation with allogeneic HSCT in this trial also resulted in an unacceptably high treatment-related mortality for Down syndrome patients, with only 33% survival at 4 years. In this study, standard timing induction followed by consolidation with chemotherapy was clearly the superior treatment for Down syndrome patients, with a 4-year DFS of 88% compared with a DFS of only 42% for this regimen in the non–Down syndrome patients. Given these collective data, COG trial A2971 used a less intensive approach for Down syndrome patients, with four cycles of standard timing low-dose cytarabine, daunorubicin, and thioguanine, followed by a cycle of high-dose cytarabine with asparaginase and then intrathecal therapy. Five-year outcome data were comparable to those of CCG2891 with OS of 84% and EFS of 79%.
The first toxicity to which DS-AML patients were identified as being particularly susceptible was cardiotoxicity. On the POG9421 AML study in which the total cumulative anthracycline equivalent dose was 535 mg/m 2 , 21% of DS children developed congestive heart failure with diminished shortening fractions on echocardiogram requiring chronic diuretics and/or inotropes. Sixty percent of these patients had congenital heart defects, and a third of the patients with cardiac dysfunction died. On both CCG2891 and A2971, the cumulative anthracycline dose was reduced to 350 mg/m 2 , but together with the overall reduction in chemotherapy on A2971, grade 3 or higher cardiac toxicity occurred in only 4% of patients during induction and 2% of patients during intensification. The most recent COG Down syndrome AML study, AAML0431, was designed to further reduce the cumulative dose of anthracycline based on A2971 and gave a cumulative dose of only 240 mg/m 2 . The study closed in December 2011 and to date no significant cardiac toxicity has been reported (J. Taub, personal communication, June 2014). However, the second cycle of induction chemotherapy during which high-dose cytarabine is given resulted in the greatest number of adverse events, including longest median duration, longest period of myelosuppression, and highest rate of bacterial infections. These toxicities, together with prior data demonstrating the exquisite sensitivity of Down syndrome AMKL blasts to cytarabine, have prompted plans for elimination of high-dose cytarabine for the majority of patients on the upcoming COG Down syndrome AML trial.
Despite the overall favorable outcomes for AML in Down syndrome, 15% to 25% of patients do not achieve long-term survival with current treatment protocols, predominantly owing to relapse or refractory disease. Lack of response to therapy is associated with very poor outcomes with survival rates of 20-26% even after HSCT. Prognostic markers in AML in Down syndrome are few because many of the predictive genetic lesions discussed earlier for non–Down syndrome AML are very infrequent findings in Down syndrome patients. To date, age older than 4 years and monosomy 7 appear the only known high-risk factors, both of which are relatively rare, and the outcome in patients harboring a monosomy 7 is still somewhat controversial. In the BFM-93 trial, 3 of 4 patients older than 4 years lacked the typical AMKL of Down syndrome and only 1 of the 3 survived. In a combined analysis of European trials, 17 of 317 Down syndrome patients diagnosed with AML were older than 4 years. DNA was available from blasts of 10 of these patients and, of these, 3 children younger than 7 years had the characteristic AMKL and 1 additional younger child had M0 AML with a GATA1 mutation. Of the 6 remaining patients without AMKL or a GATA1 mutation, all were 7 years of age or older and 4 experienced relapse. In CCG 2891, children older than 4 years of age ( n = 9) had a significantly lower EFS at 6 years of only 33% ± 31%. On A2971, patients younger than 4 years of age fared significantly better than older children (5 year EFS, 81% ± 7% vs. 33% ± 38%), although only 6 children older than age 4 years were included. An international retrospective study of 451 Down syndrome patients with AML found monosomy 7 was associated with a moderately worse outcome, with a 5-year EFS of 69%. CCG studies 2891 and A2971, as well as a recent Japanese study, found that only 11 of 16 patients with Down syndrome and AML and monosomy 7 were in complete clinical remission. These limited data suggest that older children with Down syndrome and AML, particularly given the absence of GATA1 mutations, have disease more reminiscent of non–Down syndrome AML and should be treated more aggressively. Similarly, the presence of monosomy 7 may contribute to an inferior outcome and justify more intensive therapy for this cohort. However, most children with Down syndrome and AML are younger than age 4 years and lack monosomy 7. For this group, similar to non–Down syndrome AML, MRD after induction may enable the ability to identify high-risk patients. These data will be forthcoming from the recently closed AAML0431 study and will inform future clinical trials.
Myelodysplastic Syndromes
The myelodysplastic syndromes are a heterogeneous group of clonal hematologic disorders characterized by ineffective hematopoiesis, progressive cytopenias, dysplastic transformation of hematopoietic cells, and a propensity for transformation into AML. MDS is most common in patients older than 60 years, but at the same time it is an unusual but clinically important disease of childhood.
Diagnosis and Classification
Morphology
The morphologic features of MDS include bone marrow dysplastic changes involving all three cell lineages (i.e., trilineage dysplasia). A common theme underlying these features is asynchrony between the usual cytoplasmic and nuclear differentiation programs, so that nuclei appear abnormal and less mature than the features of the surrounding cytoplasm. Typical erythrocytic abnormalities include megaloblastoid changes, perinuclear ringlike deposits of iron (i.e., ringed sideroblasts), multinucleation, nuclear budding and fragmentation, and an increased percentage of immature forms. Dysmegakaryopoiesis is characterized by micromegakaryocytes, abnormal nuclei, and abnormal nuclear lobulation. The dysplastic features of the granulocytic cells comprise immature forms, hypogranulation, and the Pelger-Huët–type abnormality. Monocytic dysplasia includes increased numbers of bone marrow monocytes, abnormal granulation with increased azurophilic granules, hemophagocytosis, abnormal nuclei, and giant forms. Bone marrow cellularity is usually normal or increased, and the reticulin concentration is increased in most cases.
Patients with MDS may present initially with a single peripheral blood cytopenia, which usually progresses to pancytopenia with anemia, leukopenia, and thrombocytopenia. The anemia in MDS is caused by ineffective erythropoiesis with low reticulocyte counts and macrocytosis, teardrop cell formation, and moderate poikilocytosis. In addition, megaloblastoid circulating nucleated red blood cells are frequently detected. Peripheral blood dysgranulopoiesis manifests as circulating myeloblasts, progranulocytes, and Pelger-Huët cells. Thus early signs may be a falling hematocrit with increasing mean corpuscular volume or unexplained thrombocytopenia. The peripheral blood may also exhibit increased numbers of monocytes and monoblasts.
Cytogenetics
Cytogenetic abnormalities are common in children with MDS and were reported in 59% of 227 children tested in one study. Chromosomal deletions are the hallmark of MDS and frequently involve the long arms of chromosomes 5, 7, and 20 ( Fig. 51-8 ). Other common cytogenetic abnormalities include trisomy 8, del(17p), +21, inv(1), and t(7;16). These chromosomal abnormalities are accompanied by other complex karyotypic changes in many cases. Other abnormalities that have been reported in pediatric cases of MDS include +6, +9, and +11, whereas deletions of chromosomes 11, 12, and 13 and Y are rare. Common balanced chromosomal translocations found in AML, such as t(8;21), t(15;17), and inv(16), are not usually detected in MDS.
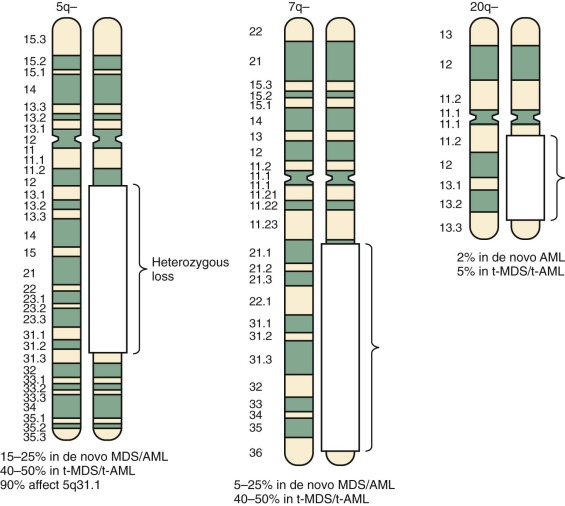
Classification
In 1982, the FAB cooperative group defined five categories for the adult myelodysplastic and myeloproliferative syndromes—refractory anemia (RA), RA with ringed sideroblasts (RARS), RA with excess blasts (RAEB), RA with excess blasts in transformation (RAEB-T), and chronic myelomonocytic leukemia (CMML). Classification of childhood MDS into defined FAB subcategories is not always straightforward. Many children show overlapping features of MPD and MDS, particularly patients with JMML and infant monosomy 7 syndrome. The classification of pediatric MDS is also complicated by the fact that some children develop MDS in the context of inherited predispositions such as neurofibromatosis type 1, severe congenital neutropenia, and Down syndrome. Using the FAB system of classification, the most common subtypes of MDS in children are RAEB and RAEB-T, whereas RA and RARS are uncommon and CMML is rare.
The WHO proposed a revised classification of MDS in 1997. The WHO system includes the following changes to the FAB criteria: elimination of the RAEB-T subtype through a reduction in the marrow blast count to 20% for the diagnosis of AML and the division of the RAEB subtype into RAEB-I, consisting of 5% to 10% marrow blasts, and RAEB-II, comprising 11% to 20% blasts. Other WHO recommendations include two new categories for patients with refractory cytopenias with multilineage dysplasia and a new category for unclassified MDS. In the new system, CMML is removed completely and reclassified as a myeloproliferative disorder.
A third classification system is used in parallel with the FAB and WHO systems, the IPSS Scoring System. This system assigns a numeric value to each of the number of cytopenias, cytogenetic abnormalities, and percentage of bone marrow blasts, and a higher score is associated with a poor prognosis. The use of the IPSS in children is of somewhat limited value. The only factors found to be predictive were bone marrow blasts greater than 5% and platelet counts greater than 100,000/µL.
Pathophysiology
Evidence for clonality in MDS comes primarily from nonrandom X-inactivation studies (see earlier) performed on the bone marrow cells of female patients with MDS. These studies have demonstrated clonal involvement of hematopoietic cells in this disorder. The trilineage dysplasia that characterizes MDS strongly suggests that the cell of origin resides within the compartment containing early hematopoietic progenitors or stem cells with multilineage potential. Early mutations in stem cells may cause a failure in myeloid cell maturation, as well as enhanced apoptosis, leading to dysplasia, whereas subsequent defects affecting myeloid cell proliferation and the development of antiapoptotic signals may cause the clonal expansion of aberrant cells and frank leukemia. An increased level of apoptosis has been documented in early MDS, which is followed by decreased blast cell apoptosis in patients whose disease transforms to AML. MDS cells have been found to overexpress certain cell surface death receptors and their ligands, which may be induced as a result of signals from inflammatory cells present in the hematopoietic microenvironment. Patients with more advanced MDS (FAB RAEB) have decreased levels of death receptor expression compared with early-stage MDS and increased expression levels of antiapoptotic factors, such as BCL2.
The most common chromosomal abnormalities in childhood MDS involve large deletions, but little is known about the genes contained in the deleted segment and whether these large genetic aberrations are inciting events leading to the development of MDS or represent secondary events. Ebert and colleagues used an RNA interference screen to identify a putative gene deleted from the distal end of the long arm of chromosome 5 in 5q− syndrome, a subtype of MDS commonly encountered in older women but uncommon in pediatric patients. They found that haploinsufficiency of RPS14 , a gene that codes for a component of the 40S ribosomal subunit, results in the characteristic phenotype of selectively diminished erythropoiesis, with preservation of megakaryocyte maturation. Moreover, the introduction of a lentiviral vector expressing RPS14 into the CD34+ cells of MDS patients demonstrates restored erythroid differentiation exclusively in those with a 5q− genotype. These elegant studies convincingly identified a specific genetic lesion underlying the 5q− syndrome and also established this ribosomal protein as critical in erythroid development. This finding is in keeping with the elucidated role of other ribosomal proteins in a number of bone marrow failure syndromes, such as Diamond-Blackfan anemia.
5q− syndrome has a reasonably favorable outcome because of its natural history of slower progression to AML and sensitivity to treatment with lenalidomide, an analogue of thalidomide. In this select population, lenalidomide (Revlimid) reduces the need for red cell transfusions and induces significant cytogenetic responses, which has resulted in its recent approval by the FDA for the treatment of MDS patients with 5q− syndrome. Although this therapy has demonstrated impressive efficacy in this condition, deletion of this distal region of 5q is relatively rare. Only a minority of MDS patients without this cytogenetic abnormality respond to lenalidomide, and responders possess no distinguishing clinical or laboratory characteristics compared with nonresponders. However, other studies suggest that responders have a unique genetic signature consisting of low expression levels of genes typically involved in terminal erythroid differentiation. These results imply that the restoration of normal red cell development underlies lenalidomide’s mechanism of action, particularly in non-5q−, whereas in 5q− syndrome, lenalidomide may more directly suppress the malignant clone. Clinically, this suggests that genetic profiling could be used to select a group of MDS patients without 5q− deletions who would be more likely to respond to this agent.
These large deletions also likely contain certain important tumor suppressor genes, the elimination of which contributes to MDS pathogenesis. For example, we completed a detailed analysis of the more proximally deleted region of the long arm of chromosome 5 and identified α-catenin as a novel tumor suppressor gene in MDS. In both primary patient samples and AML cell lines with a deletion of 5q−, α-catenin expression levels were found to be specifically and substantially decreased through the combination of a deletion on one chromosome and epigenetic silencing by hypermethylation and histone deacetylation of the promoter of the second allele. Another group has identified the WT1 transcription factor, early growth response 1 (EGR1) , as a putative candidate gene in this same region and demonstrated that haploinsufficiency of this tumor suppressor gene can result in MDS or MPD after treatment with an alkylating agent.
In addition to gene deletions, a number of other genes have been found to contribute to MDS pathogenesis. Balanced translocations, such as those seen in de novo AML, are uncommon in MDS, but some of the same genes may be involved. Other genetic lesions appear to be specific for MDS. We and others have shown that certain HOX genes, most notably HOXA9 , are overexpressed in the HSCs of patients with MDS. Translocations involving HOX genes and the nucleoporin gene (NUP98) on chromosome 11, including NUP98-HOXA9 and NUP98-HOXA13, have been found in rare patients with MDS. Certain genetic abnormalities are more characteristic after prior treatment with chemotherapy. Overexpression of the EVI1 transcription factor on chromosome 3, either through gene rearrangements or rare translocations, has been associated with MDS after chemotherapy. Similarly, nucleophosmin and myeloid leukemia factor 1 (NPM-MLF1) fusion gene, formed as a result of a t(3;5) chromosomal translocation, occurs in rare cases of MDS or AML arising from MDS. Mutations in RAS and FLT3 , such as those seen in de novo AML, are found in patients with MDS, particularly those with 7q− and often after treatment with alkylating agents. These patients may also have mutations in the AML1 gene, whereas the AML1-ETO translocation is rare in MDS. Prior treatment with alkylators can also lead to MDS with 5q−, and these patients often have a mutation in TP53 , a gene that is also frequently mutated in AML cases with a complex aberrant karyotype, which include a 5q deletion, but rarely in classic de novo AML.
More recently, owing to responses in MDS to epigenetic regulating agents, such as DNA methyltransferase inhibitors, and the identification of a number of mutations in epigenetic regulating genes in AML, a series of studies using high-throughput genomic approaches have identified similar abnormalities in MDS. Mutations in TET2 occur in approximately 25% of adults with MDS, whereas DNMT3A and IDH1 and IDH2 mutations were found in 3% to 10% of patients. In addition, mutations have been found in histone modifying enzymes, including EZH2 , which is rare in de novo AML, and ASXL1 , both associated with a worse outcome in adult MDS. Finally, novel mutations in genes involved in the assembly of the spliceosome that removes introns from transcribed pre-mRNA have been identified. The genes most commonly involved include SF3B1 and U2AF1 . However, MDS is much rarer in children, and all of these mutations appear to be rare in pediatric MDS; thus their impact on pathogenesis and prognosis remain uncertain at this time.
Inherited and Environmental Predisposing Factors
Several environmental exposures and inherited conditions are associated with an increased risk for MDS and AML. As noted, alkylating agents and topoisomerase II inhibitors can predispose to MDS and AML, which can occur as treatment-related second malignancies in patients treated for ALL and many other types of cancer. Secondary disease associated with alkylating agent chemotherapy tends to occur from 3 to 11 years after treatment, after which the incidence appears to plateau. The disease often presents as MDS, and an underlying predisposition may involve defective DNA repair mechanisms. Commonly, there are deletions of chromosomes 5 or 7. MLL gene rearrangements are often found in AML cases that arise after treatment with topoisomerase II inhibitors such as etoposide, but these patients tend to present with frank AML after a short latency of 1 to 3 years from therapy and without an MDS prodrome.
A number of genetic conditions are associated with an increased risk for MDS and subsequent AML, particularly the bone marrow failure syndromes and chromosomal breakage disorders. Some disorders, such as Fanconi anemia, in which patients demonstrate both pancytopenia and increased sensitivity to DNA damage, suggest that impaired DNA repair may be a mechanism for malignant transformation. However, other marrow failure syndromes are not characterized by increased susceptibility to chromosomal breakage such as dyskeratosis congenita or may present predominantly as a single cytopenia, such as neutropenia in Shwachman-Diamond syndrome, anemia in Diamond-Blackfan anemia, and low platelets in amegakaryocytic thrombocytopenia. The evolution of MDS in these conditions is variable (10% to 20% in Shwachman-Diamond syndrome compared with 1% in Diamond-Blackfan anemia), and the mechanism remains elusive, although defective ribosomal processing may be involved, given the identified functions of the target proteins involved in a number of these conditions. In addition, there are several MDS/AML predisposition genes that have been identified, including loss-of-function alterations affecting the key hematopoietic transcription factor genes, RUNX1, CEBPA, and, most recently, GATA2 . Germline mutations in RUNX1 can result in familial platelet disorder with a propensity to myeloid malignancy (FPD/AML). These patients have thrombocytopenia or normal platelet numbers but abnormal platelet function and can evolve into MDS or AML. A recently identified related disorder termed thrombocytopenia type 2 has been linked to mutations in a gene called ANKRD26 at 10p11.2-p12 in a number of Italian and American families with a progression to frank myeloid and lymphoid malignancies, with an MDS prodrome rarely described. Deletions of the granulocyte-specifying gene CEPBA have been described in a small number of families resulting in an FAB M1 or M2 subtype AML; however, an MDS prodrome has not been described. The CEBPA mutations are autosomal dominant, similar to those involving RUNX1 , but fully penetrant, in contrast to the highly variable penetrance observed for RUNX1 lesions. Loss-of-function mutations or deletions involving GATA2 , which plays a role in early blood, vascular, and lymphatic development, have been described as a cause of a familial MDS/AML syndrome. These patients may present with neutropenia, often with an associated monocytopenia or with so-called monoMAC syndrome/DCML deficiency associated with monocytopenia and dendritic, B-cell, and NK-cell lymphopenias with a predisposition to nontuberculosis mycobacterial infections. Some of these patients may also develop lymphedema.
Clinical Presentation
Children with MDS generally present with the signs and symptoms associated with bone marrow failure. Abnormalities in the peripheral blood may also be detected on routine medical examinations in asymptomatic children. Symptomatic children often present with some degree of fatigue, fever, malaise, or infections. Physical examination frequently reveals pallor, easy bruising, and petechiae. Splenomegaly, hepatomegaly, and lymphadenopathy are uncommon findings. The peripheral blood cell counts usually reveal anemia, neutropenia, and thrombocytopenia.
Differential Diagnosis
With a careful history and physical examination, as well as the evaluation of peripheral blood and a bone marrow aspirate, biopsy, and cytogenetics, a diagnosis of MDS can be made. Severe nutritional deficiencies of vitamin B 12 and folate should be excluded, because they may impart a megaloblastic appearance to the bone marrow. Other nutritional deficiencies such as thiamine, pyridoxine, iron, and riboflavin can also present as some degree of bone marrow dysplasia and should be considered in the differential diagnosis. Occasionally, severe aplastic anemia can be difficult to distinguish from MDS, although a bone marrow biopsy should resolve this question. Certain viral infections (e.g., human immunodeficiency virus, parvovirus B19, Epstein-Barr virus, human herpesvirus 6, cytomegalovirus) and exposures (e.g., irradiation, chemotherapy, organic solvents) are associated with bone marrow failure and should be considered before making the diagnosis of MDS. The bone marrow cell karyotype should be determined to identify any clonal abnormalities. In addition, evidence has suggested that whole-genome scan by SNP analysis may detect clonal abnormalities in some patients with a normal bone marrow cell karyotype.
Treatment
The rarity and heterogeneity of MDS in children, the lack of consensus on nomenclature, and the need for a risk-based classification system specific for pediatric MDS have hindered the development of appropriate therapy for pediatric MDS. Most clinical trials studying MDS have enrolled adults, in whom the disease may be different, and there have been few meaningful trials enrolling children. Regardless of treatment, the overall clinical outcome of children with MDS is guarded.
The only proven curative treatment for MDS is allogeneic HSCT. AML-based chemotherapy without HSCT has resulted in poor survival in the 30% range. Prior to HSCT, or if an appropriate donor was not available, treatment primarily consisted of supportive care with packed red blood cell or platelet transfusions, parenteral antibiotics, and hydration. Other noncytotoxic therapies, including hormone therapy (e.g., androgens, glucocorticoids), recombinant hematopoietic growth factors (e.g., G-CSF, GM-CSF, erythropoietin), and differentiating agents (e.g., 13- cis -retinoic acid, ATRA), have been used in adults and have shown minimal benefit. Low-dose chemotherapy has been used in an attempt to promote differentiation of the malignant hematopoietic clone. Although studies using low-dose cytarabine have shown improvement in peripheral counts and decreased marrow blast percentages in some patients with MDS, the responses have not been sustained. Other agents, such as melphalan, hydroxyurea, etoposide, topotecan, 6-mercaptopurine, and busulfan, have been used with only partial or temporary responses in small numbers of patients.
A number of new agents have emerged for the treatment of pediatric MDS after randomized clinical trials in adult patients. These therapies have evolved out of a growing interest in the field of epigenetics and the concept that the repression of key tumor suppressor genes in MDS may occur because of aberrant methylation and histone deacetylation, instead of or in concert with gene deletions and mutations. Studies involving the CDKN2B (p15 INK4B ) tumor suppressor gene have demonstrated evidence of methylated CpG islands in the promoter region, preventing gene transcription and leading to gene repression. Since these initial studies, a number of other genes have been found to have reduced expression because of DNA methylation, thus prompting clinical trials of demethylating agents in MDS. Based on the impressive response rates and delayed progression to AML compared with supportive care, two demethylating agents, 5-azacytidine and decitabine, have been approved by the FDA for the treatment of MDS in adults. These agents have shown the greatest efficacy in low-risk MDS. They are given in a low-dosage schedule, and patients may need to be treated for several months before a clinical response is achieved. Although these agents have improved the time to progression in adult MDS, they cannot cure the disease. The role of these therapies in pediatric MDS is currently under active investigation. As indicated earlier, lenalidomide is now approved for the treatment of patients with MDS with the 5q− cytogenetic abnormality and appears to work best in those whose MDS clone lacks any other cytogenetic abnormalites.
For children with MDS, allogeneic HSCT remains the treatment of choice. In the absence of a sibling donor, a matched unrelated donor should be sought. Children with lower-risk MDS are frequently treated with allogeneic HSCT without prior chemotherapy, particularly if a matched sibling is available, to avoid the need for large numbers of transfusions. In more advanced MDS (WHO RAEB1 and RAEB2), AML-type chemotherapy has been often administered to induce a remission or to reduce the disease burden before HSCT. A study from the Fred Hutchinson Cancer Research Center in Seattle in children with MDS demonstrated a 3-year survival rate of patients with lower-stage MDS of 74% compared with 68% for patients with more advanced disease. The European Working Group on MDS in Children (EWOG-MDS) reported a 55% 5-year EFS rate for 33 pediatric patients with MDS after HSCT. A recent pediatric study from the University of Minnesota demonstrated an OS of 53% and a DFS of 48% at 3 years for 37 pediatric patients with MDS who received allogeneic HSCT after myeloablative conditioning over a 20-year period ending in 2010. Earlier HSCT without preconditioning chemotherapy was associated with an improved outcome, suggesting that delaying transplant to reduce disease burden with AML-directed therapy is not warranted if an appropriate donor is available. These results are in keeping with those found in a European study of pediatric MDS patients who received a myeloablative allogeneic HSCT in which the OS was 63% at 5 years. This study similarly showed no benefit to pre-HSCT chemotherapy except for the group who had more advanced myelodysplasia-related AML, a subgroup not included in the prior study. A longer interval between diagnosis and HSCT was actually associated with less relapse but may have been confounded by the number of lower-risk MDS patients in this group. TRM during transplant can be a significant issue, resulting in some groups attempting reduced-intensity conditioning regimens. These approaches have had variable success, with some studies reporting lower DFS rates.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


