Chapter Outline
EMERGENCIES IN PATIENTS WITH NEWLY DIAGNOSED LYMPHOMA
Epidemiology and Causative Factors
Initial Evaluation and Staging
Anaplastic Large-Cell Lymphoma
Rare Lymphomas in Children and Adolescents
POSTTRANSPLANTATION LYMPHOPROLIFERATIVE DISEASE
Lymphomas are neoplasms caused by the malignant transformation of the constituent cells of the immune system. Combined, Hodgkin and non-Hodgkin lymphomas are the third most common malignancies in children and adolescents, with Hodgkin lymphoma being the most common cancer in people between the ages of 15 and 18 years.
The treatment of children and adolescents with lymphoma is one of the important success stories in pediatric oncology. The transformation of what were uniformly fatal diseases to those in which cure is the expectation occurred first for those with Hodgkin lymphoma and more recently for the majority of children with non-Hodgkin lymphoma. The understanding of normal and abnormal lymphoid biology has been critical to the develop systems for classification and for the evolution of disease-specific therapy.
This chapter reviews the clinical and pathologic features, including molecular and cellular biology, of the most common pediatric lymphoma subtypes and reviews the current strategies for evaluation and treatment.
Lymphadenopathy
The child with lymphadenopathy poses a relatively common diagnostic challenge for the pediatrician. Over one third of 5-year-old children seen for well-child care and two thirds evaluated for a sick visit have palpable adenopathy. In most children, lymph-node enlargement results from transient, self-limited infectious processes that resolve without sequelae. However, serious, life-threatening benign and malignant diseases may present with lymphadenopathy as the first manifestation. Thoughtful decision making is critical regarding further investigation, including which children require biopsy of their nodes for pathologic evaluation.
Various definitions of adenopathy have been described, including specific measurements in the radiologic literature. In general, adenopathy is defined as any node greater than 1 cm in dimension. Epitrochlear nodes are considered enlarged if they are greater than 0.5 cm, and inguinal nodes are enlarged if they are greater than 1.5 cm. Lymphadenopathy is considered localized if it involves a single node or single nodal area and generalized if it involves more than 2 noncontiguous nodal groups and may include hepatosplenomegaly. Chronic adenopathy is variably defined but in general can be considered as enlargement that persists for longer than 3 weeks.
Enlargement of lymph nodes results from expansion and recruitment of normal lymph node cells, expansion of abnormal immune cells, or infiltration of extrinsic cells. Certain considerations narrow the diagnostic possibilities in evaluation of the child with lymphadenopathy. Not all masses in children that appear to be lymphadenopathy represent enlarged lymph nodes; non–lymph-node masses that may mimic cervical lymphadenopathy include thyroglossal duct cysts and branchial cleft cysts. Evidence of recent or current upper respiratory infection and the presence of tender cervical adenopathy suggest inflammation as the cause. The presence of associated systemic symptoms, the chronicity of the adenopathy, and whether the adenopathy is generalized or regional are important considerations in the differential diagnosis. In addition, the age of the patient is important in considering the differential diagnosis, especially as it relates to potential malignant conditions. An abbreviated list of common causes of lymphadenopathy is presented in Box 53-1 . More complete listings have been published elsewhere.
Infection
Bacterial: Staphylococcus aureus, group A Streptococcus, Bartonella henselae, brucellosis, tularemia
Viral: Epstein-Barr virus, cytomegalovirus, human immunodeficiency virus, measles, rubella
Fungal: Histoplasmosis, coccidioidomycosis, Cryptococcus
Protozoan: Toxoplasmosis, malaria
Mycobacterial: Tuberculosis, atypical mycobacteria
Autoimmune Disease
Juvenile rheumatoid arthritis, systemic lupus erythematosus, serum sickness, autoimmune lymphoproliferative syndrome
Storage Disease
Niemann-Pick disease, Gaucher disease
Drug Reaction
Phenytoin and others
Malignancy
Lymphoma, leukemia, metastatic solid tumors, histiocytic disorders
Miscellaneous
Sarcoidosis, Kawasaki disease, Kikuchi disease
Approach to the Patient
The management of the child with lymphadenopathy should be focused on obtaining clues to a diagnosis from the history, physical examination, and noninvasive testing, with the goals of ascertaining whether the lymphadenopathy is likely to be a manifestation of a serious illness and determining as early as possible in the workup whether the child should undergo lymph node biopsy. The history should include the duration of the adenopathy, associated symptoms, evidence of recent infection in the regions drained by the involved lymph node, exposure to illnesses, cats, or rodents, and current medications. The physical examination should be focused on ascertaining the location and number of enlarged nodes, their size, and their texture. Involvement of supraclavicular lymph nodes suggests mediastinal pathology and is usually associated with a serious disease mandating a prompt workup including a chest radiograph. In contrast the involvement of upper cervical lymph nodes is more likely to be the result of an upper respiratory tract infection.
For the child with generalized lymphadenopathy initial evaluation to be considered should include a complete blood count and a chest radiograph. Testing for possible infectious pathogens including viruses, fungi, and bacterial and mycobacterial agents should be considered. If no clues emerge, lymph node biopsy is likely indicated.
The workup of a child with localized or regional lymphadenopathy must be tailored to the individual child. Retrospective studies have attempted to identify those children with adenopathy who are more likely to have an underlying malignancy. Of children who underwent node biopsies, predictive factors for the etiology being malignancy included age older than 10 years, node size greater than 2.5 cm, and supraclavicular site. Additional factors that were possibly associated included persistence of the node for longer than 6 weeks, having the node be “fixed” by palpation, and having more than one nodal area involved. In asymptomatic children whose adenopathy does not have high risk features, observation with careful measurement of lymph node size and possibly an empirical trial of antibiotic therapy is a reasonable strategy. If the nodes increase in size or fail to decrease to normal size after several weeks of observation, a lymph-node biopsy should be considered.
Masses occurring in the mediastinum represent urgent and challenging diagnostic problems, whether the result of enlarged lymph nodes or of the involvement of extralymphatic tissues. Lymphomas account for a significant portion of anterior and middle mediastinal masses in children and adolescents. Their proximity to vital structures and their propensity to cause life-threatening symptoms from vascular or airway compromise mandate an expeditious, systematic approach involving close cooperation among the surgeon, radiologist, oncologist, anesthesiologist, radiotherapist, and pathologist. A differential diagnosis of mediastinal masses according to location within the mediastinum is shown in Box 53-2 .
Emergencies in Patients with Newly Diagnosed Lymphoma
Children with lymphoma can have life-threatening findings such as respiratory distress and superior vena cava (SVC) syndrome caused by a mediastinal tumor, bowel obstruction from large abdominal masses, cranial nerve palsies, and paraplegia in the case of central nervous system (CNS) involvement and, in rare cases, disseminated intravascular coagulation. Patients may exhibit the metabolic derangements of acute tumor lysis syndrome (ATLS), although more often this problem arises after the initiation of therapy. Invasive diagnostic measures, especially those requiring anesthesia, can create emergencies for children with lymphomas.
A mediastinal tumor can cause respiratory distress as a result of tracheal or other large-airway compression. Patients often have cough, shortness of breath, and orthopnea. If there is considerable respiratory impairment, then all invasive diagnostic procedures need to be completed expeditiously. In these patients general anesthesia carries potential for substantial risks including inability to ventilate the child because of airway compression as well as swelling of the tracheal mucosa secondary to intubation, which may lead to worsening of critical constriction. Diagnostic procedures often need to be completed with local anesthesia alone. The least invasive procedure that will allow for attainment of diagnostic tissue should be undertaken. In emergent situations, completion of staging (e.g., diagnostic lumbar puncture) may need to be deferred. Once tissue has been obtained, then initiation of steroids can be considered until pathologic diagnosis is complete. Very rarely for the patient with impending respiratory failure empirical therapy without specific diagnosis may needed. Cytoreduction with prednisone 60 mg/m 2 /day and cyclophosphamide 100 to 200 mg/m 2 /day or mediastinal radiation with doses of 10 to 15 Gy may provide a life-saving intervention.
In addition to respiratory impairment, mediastinal tumors can cause SVC syndrome by compression of the SVC and subsequent venous congestion. Patients have dilated neck veins, facial swelling, and discoloration. The life-threatening risk is from CNS venous strokes, often presenting as confusion or somnolence and less often as seizures or focal neurologic deficits. The tempo of progression of SVC syndrome in children tends to be much quicker in children than in adults as a consequence of more rapidly growing tumors. Again, in these patients expeditious evaluation and initiation of therapy is of critical importance.
Patients who have cranial nerve palsies and especially those with signs of incipient paraplegia also require urgent care. As in other pediatric malignancies, interventions may include surgical decompression, radiation, and chemotherapy. In general for children with lymphomas, which tend to be highly and quickly responsive to chemotherapy, initiation of therapy with prednisone or dexamethasone with or without cyclophosphamide is often the best approach. Clinicians should be aware that lumbar puncture in patients with bulky CNS disease may carry the risk of brainstem herniation. Urgent imaging should be considered before the procedure.
ATLS describes the metabolic derangements that occur with tumor-cell breakdown and is characterized by various combinations of hyperuricemia, hyperkalemia, and hyperphosphatemia with or without hypocalcemia and can lead to seizures, arrhythmias, renal failure, and death. ATLS can be present before the initiation of treatment but more commonly occurs immediately after the initiation of chemotherapy. The incidence of ATLS differs among tumor types and depends primarily on the growth fraction, tumor mass, and chemotherapeutic sensitivity of the tumor. Adult and pediatric risk stratification algorithms for ATLS syndrome has been made and includes age, malignancy type, stage/lactate dehydrogenase (LDH) level, and renal function as predictive factors. Patients with non-Hodgkin lymphoma (NHL) are at high risk, particularly those with advanced stage Burkitt lymphoma (BL), Burkitt leukemia, or lymphoblastic lymphoma (LL) with LDH greater than twice normal.
Vigorous hydration, alkalinization, and the administration of allopurinol, an inhibitor of xanthine oxidase, have been considered mainstays of prevention and treatment of ATLS and are aimed at the reduction of uric acid production and prevention of its precipitation in the kidneys. Evidence supporting the use of alkalization is controversial, and some contemporary guidelines do not recommend its use. For patients considered at low risk, no specific intervention is required. For those at intermediate risk, careful monitoring of fluid and electrolytes is critical. Hyperhydration and allopurinol are almost always sufficient.
For those with higher risk for tumor lysis or who have evidence of the syndrome before the initiation of therapy, rasburicase should be considered. This agent is a recombinant urate oxidase that catalyzes the conversion of uric acid to allantoin. Allantoin is an inactive metabolite that is five to ten times more soluble than uric acid, so renal excretion is facilitated. A single dose will result in the rapid reduction in uric acid levels in patients with hyperurecemia. It is contraindicated in patients with a history of glucose-6-phosphate dehydrogenase (G6PD) deficiency because of substantial risks of precipitating severe hemolysis. In a systematic review of published randomized trials, rasburicase was highly effective at reducing elevated uric acid, although the impact on mortality and renal failure was unclear. In a French study of children with advanced stage B-cell NHL that incorporated the use of rasburicase, the incidence of need for dialysis during the first days of treatment was only 2.6% compared with 16% and 23% in other cooperative studies using the same French protocol that used only allopurinol.
In some patients, even with optimal ATLS care renal function is insufficient and early hemodialysis should be instituted. This situation may occur because of direct infiltration of the kidneys, obstruction of the urinary tract caused by lymphomatous compression, established urate or calcium phosphate nephropathy, or a combination of these conditions.
Hodgkin Lymphoma
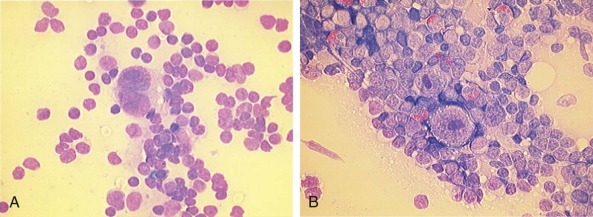
Hodgkin lymphoma was first described in 1832 by Thomas Hodgkin as a disorder characterized by a peculiar enlargement of the absorbent (lymphatic) glands and spleen and was named in 1865 by Sir Samuel Wilks. Sternberg and Reed, in 1898 and 1902, respectively, are credited with the first definitive and thorough description of the binucleate or multinucleated giant cells that when present, are considered pathognomonic of this disorder ( Fig. 53-1 ). The malignant nature of the disease was proven when Seif and Spriggs confirmed the clonal origin of the malignant cell by cytogenetic analysis.
Before 1960 Hodgkin lymphoma was an almost uniformly fatal disease. In the 1960s investigators at Stanford and elsewhere developed the use of megavoltage radiotherapy and defined treatment fields including mantle, inverted Y, total nodal, and total lymphoid irradiation. Subsequently in the late 1960s and early 1970s the first randomized trials were initiated. Concurrently chemotherapy agents were being investigated. After the observation of the lympholytic effect of nitrogen mustard, this and other agents were used individually in the attempt to treat patients with leukemia and lymphoma. A major step forward toward the use of chemotherapy for Hodgkin lymphoma was the introduction of the four-drug combination of mechlorethamine, vincristine (Oncovin), procarbazine, and prednisone (the MOPP regimen), which led to substantial rates of cure in previously incurable patients.
Over the next 20 years, standard therapy included clinical and surgical staging including laparotomy and splenectomy, as well as extended field radiation with doses of 36 to 44 Gy with or without combination chemotherapy. The probability of cure in children and adolescents using this strategy approached 90%. Early on, however, late effects of therapy were noted. The first to be observed was the impact on musculoskeletal growth of young children receiving high-dose extended field radiation. Subsequently, the long list of therapy-related late effects emerged to include second malignancies, serious cardiac and pulmonary compromise, infectious complications, and sterility. For the last two decades the focus of the development of care for patients with Hodgkin lymphoma has been to maximize the probability of long-term, disease-free survival but to do so while minimizing risks of late effects.
Unlike the complex evolution of classification systems for NHL, the systems used for Hodgkin lymphoma have been more straightforward. The Rye modification of the Lukes-Butler system of classification of Hodgkin disease was universally accepted for 25 years and formed the basis for the current World Health Organization (WHO) classification. Studies of the biology and clinical course of the disease have shown that Hodgkin lymphoma should be subdivided into two entities, classic Hodgkin lymphoma (CHL) and nodular lymphocyte–predominant Hodgkin lymphoma (NLPHL) ( Table 53-1 ). The two entities will be discussed separately in the next sections.
| CHL | NLPHL | |
|---|---|---|
| Percentage of patients | 92%-95% | 5%-8% |
| Histologic hallmark | Reed-Sternberg cell | Lymphocyte-predominant cells (also called popcorn cells ) |
| Immunophenotype | CD15 and CD30 common, CD20 rare | CD15 and CD30 rare, CD20 common |
| EBV association | Yes in 40% | Negative |
| Percentage male | 50% | 75% |
| Peak incidence | 15-20 years | 13 years |
| Disease at presentation | Majority with stage II or greater; mediastinal involvement common | Majority with stage IA or IIA; mediastinal involvement uncommon |
Classic Hodgkin Lymphoma
Pathologic Features
CHL is characterized by the presence of mononuclear Hodgkin and multinucleated Reed-Sternberg cells. Hodgkin and Reed-Sternberg cells are usually in the minority, residing in a reactive infiltrate of a variable mixture of nonneoplastic lymphocytes, eosinophils, neutrophils, plasma cells, fibroblasts, and collagen fibers present in response to cytokines produced by the tumor. The Reed-Sternberg cells are large cells with abundant, slightly basophilic cytoplasm and have at least two nuclear lobes or nuclei containing a prominent inclusionlike eosinophilic nucleolus. Diagnostic Reed-Sternberg cells must have at least two nuclei in two separate lobes. Mononuclear variants are termed Hodgkin cells and often have a more intense basophilic cytoplasm. Microdissection techniques have enabled the isolation of Reed-Sternberg cells from frozen sections and the investigation of their lineage commitment. In more than 98% of cases, Reed-Sternberg cells are B cells, as defined by monoclonal immunoglobulin gene rearrangements. Only a few cases have shown clonal T-cell receptor (TCR) gene rearrangement in the Reed-Sternberg cells, suggesting T-cell origin. Reed-Sternberg cells of CHL express the B-lineage antigens CD20 and CD79a in variable proportions, whereas the B-cell–specific activator protein (BSAP), a product of the PAX5 gene, is expressed in about 90% of cases. Reed-Sternberg cells are almost invariably positive for the CD30 antigen and usually express the CD15 antigen, whereas the expression of the epithelial membrane antigen (EMA) is rare. In Epstein-Barr virus (EBV)–positive cases of CHL, the Reed-Sternberg cells express the EBV latency type II pattern latent membrane (LMP) protein 1 and Epstein-Barr nuclear antigen (EBNA) 1, but without EBNA2.
Based on the characteristics of the reactive infiltrate and the morphology of the Reed-Sternberg cells, four subtypes of classic Hodgkin lymphoma are distinguished in the WHO classification : lymphocyte-rich Hodgkin lymphoma (LRHL), lymphocyte-depleted Hodgkin lymphoma (LDHL), mixed-cellularity Hodgkin lymphoma (MCHL), and nodular sclerosis Hodgkin lymphoma (NSHL). The immunophenotypic and genetic features of the Reed-Sternberg cells are identical in these histologic subtypes, whereas they differ in clinical features and association with EBV. CHL is associated with overexpression and an abnormal pattern of cytokines and chemokines and their receptors by Reed-Sternberg cells and the cells of the reactive background. The abnormal cytokine and chemokine expression most likely accounts for the abundant admixture and pattern of inflammatory cells in CHL lesions as well as for distinct clinical features.
The NSHL subtype is characterized by a nodal growth pattern with collagen bands that surround at least one nodule, the formation of clusters of Reed-Sternberg cells, and so-called lacunar cells (mononuclear Hodgkin cells with only moderately prominent nucleoli). NSHL can be grade 1 or 2, mainly depending on the number of Reed-Sternberg cells in distinct nodules. The EBV-encoded LMP1 antigen is less commonly expressed than in other subtypes.
The MCHL subtype is characterized by scattered classic Reed-Sternberg cells in a diffuse or vaguely nodular mixed inflammatory background without sclerosis. Partial involvement of the lymph node can be observed in MCHL, described as interfollicular disease. The EBV-encoded LMP1 antigen is more commonly expressed than in NSHL.
The LRHL and LDHL subtypes are relatively rare in children. The LRHL subtype contains scattered Reed-Sternberg cells and a nodular or diffuse background of small lymphocytes but with an absence of neutrophils and eosinophils. The LDHL subtype is a diffuse form of CHL, rich in Reed-Sternberg cells and/or depleted of nonneoplastic lymphocytes and is often sarcomatoid in appearance.
In some patients, distinction between true Hodgkin lymphoma and certain subtypes of NHL can be difficult. Grey-zone lymphomas are those that do not fit into a single disease entity. The WHO classification includes the entity of “B-cell lymphoma, unclassifiable with features intermediate between diffuse large B-cell lymphoma (DLBCL) and classic Hodgkin lymphoma,” although this entity is exceptionally rare in children.
Molecular and Cellular Biology
The cell of origin of the malignant cells in CHL was long a matter of controversy because of the low incidence of Reed-Sternberg cells in the tumors and their unusual immunophenotype in that they lack expression of immunoglobulins and other B-cell markers; rather, they express markers characteristic of dendritic cells, granulocytes, monocytes, and T cells. However, the identification of immunoglobulin gene rearrangement and somatic hypermutation in microdissected Reed-Sternberg cells in almost all cases of CHL has demonstrated that these cells derive from postgerminal B cells. In addition these studies have demonstrated that about 25% of CHL cases carry crippling immunoglobulin-gene rearrangements that destroy the function of the immunoglobulin protein. These nonfunctional rearrangements typically induce rapid apoptosis in normal B cells, suggesting that Hodgkin lymphoma may originate from germinal-center B cells that have escaped apoptosis. Analysis of TCR rearrangements has shown that despite the common expression of T-cell markers, CHL originates from T cells in only 1% to 2% of cases.
In NLPHL, the malignant popcorn cells show expression of B-cell markers such as CD20 and CD79a, suggesting a B-cell origin for these tumors as well. As in CHL, the analysis of immunoglobulin genes in microdissected tumors has shown clonal and somatically mutated gene rearrangements. In addition, 50% of cases have shown evidence of ongoing somatic hypermutation, which strongly suggests that they derive from germinal center B cells.
Mechanisms of Transformation.
EBV has been associated with the pathogenesis of CHL, which shows EBV infection of the Reed-Sternberg cell in 40% of the cases in the Western world and 90% in Central America. Importantly, EBV can immortalize human B cells in vitro, and Reed-Sternberg cells are clonally infected by EBV, which suggests that viral infection occurs early in the pathogenesis of the disease. EBV positive Reed-Stenberg cells express EBNA1, LMP1, and the LMP2a viral gene, which is characteristic of the viral latency II stage. Mechanistically, LMP1 mimics an active CD40 receptor and can activate the nuclear factor κB (NF-κB) pathway, which is constitutively active in Hodgkin lymphoma (see later). LMP2, a second viral gene product, mimics B-cell receptor signaling and might play a role in rescuing Reed-Sternberg cells from apoptosis with immunoglobulin-crippling mutations. Cytogenetically, Reed-Sternberg cells often show aneuploidy and chromosomal abnormalities, with 20% of the cases harboring chromosomal translocations involving the immunoglobulin loci, a hallmark of many B-cell lymphomas.
The most prominent molecular abnormality found in Hodgkin lymphoma is the constitutive activation of the NF-κB pathway. NF-κB functions as an important survival-signaling pathway in B cells in response to the activation of members of the tumor necrosis factor (TNF) receptor family (CD30, CD40). In Hodgkin lymphoma, somatic mutations in different elements of the NF-κB pathway keep it constitutively activated. Thus the REL gene, which encodes an NF-κB component, is amplified in the of 30% of the cases. Gains of NF-κB–inducing kinase (NIK), a positive regulator of the alternative NF-κB pathway, are also commonly found in Reed-Sternberg cells, and translocations and amplifications resulting in overexpression of the BCL3 gene in Reed-Sternberg cells has been described. Moreover, mutations of the gene-encoding NF-κB inhibitors IKBA and IKBE are found in 10% to 20% of cases of Hodgkin lymphoma. Most significantly TNFAIP3, which encodes A20, a ubiquitin-modifying enzyme involved in the negative regulation of NF-κB signaling, harbors loss of function mutations and deletions in almost 45% classic Hodgkin lymphomas. Notably, TNFAIP3 mutations and deletions are more common in EBV-negative tumors and are rarely found in lymphocyte-predominant Hodgkin lymphomas. Finally, activation of TNF signaling by the microenvironment and activation of NF-κB by the LMP1 EBV oncogene may also contribute to aberrant NF-κB signaling.
Gene amplifications involving the JAK2 locus have been described in about 20% of CHL cases, suggesting a role for JAK2 activation in the pathogenesis of this disease. In addition, rearrangements of JAK2 including a recurrent SEC31A–JAK2 translocation have been described in rare cases of Hodgkin lymphoma. Moreover, loss of function mutations in SOCS1 , a negative regulator of Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling are present in about 50% of classic and lymphocyte-predominant Hodgkin lymphomas and are associated with increased levels of JAK signaling.
Translocations involving the major histocompatibility complex (MHC) class II transactivator gene CIITA , resulting in impaired MHC class II expression, are found in 15% of CHL cases and seem to be associated with unfavorable prognosis. In contrast to CHL, the malignant cells of the lymphocyte-predominant type of Hodgkin lymphomas are always EBV-negative, and there is little information on the molecular lesions implicated in the pathogenesis of this group. Chromosomal translocations involving the BCL6 oncogene can be detected in 30% of cases. BCL6 is a zinc-finger transcription repressor that functions as a master regulator of germinal-center formation and drives B-cell transformation. Thus Bcl6 -null mice fail to generate germinal centers in response to immunization, and deregulated expression of Bcl6 induces B-cell lymphomas in mice. Mechanistically, aberrant expression of BCL6 in lymphoma hijacks the role of BCL6 in the control of B-cell activation, differentiation, deoxyribonucleic acid (DNA) damage response, cell-cycle arrest, and apoptosis. Finally, about 40% of lymphocyte-predominant Hodgkin lymphoma tumors show mutations in SOCS1, but mutations in TNAIP3 and NFKBIA are rare despite strong NF-κB activity.
Cellular Microenvironment: Cytokines and Chemokines.
The cellular and cytokine microenvironment surrounding the lymphoid cells in Hodgkin lymphoma plays an essential role in the pathogenesis of this disease. In particular, the expression of soluble factors and their receptors by malignant cells and the reactive microenvironment seems not only to mediate the inflammatory characteristics observed in the histology of Hodgkin lymphoma, but also to contribute to the proliferation and survival of the malignant clone. Thus the cellular microenvironment supports and is supported by a network of cytokines secreted in autocrine and paracrine loops that are essential for the proliferation of Reed-Sternberg cells and the maintenence of a favorable inflammatory environment rich in regulatory T cells (Tregs) and eosinophils.
Both interleukin (IL) 13 and the IL-13 receptor IL-13RA1 are expressed in Hodgkin lymphoma and constitute an important autocrine loop for Reed-Sternberg cells. Other cytokines expressed in Hodgkin lymphoma and thought to influence survival of these cells include IL-4, IL-6, IL-7, IL-9, and IL-15.
The vast majority of cells in a tumor biopsy of Hodgkin lymphoma are not malignant cells but represent an inflammatory-like cellular infiltrate composed mainly of CD4+ T lymphocytes intermixed with macrophages, eosinophils, plasma cells, and fibroblasts. Most lymphocytes present in this infiltrate are Tregs, which play an important role in protecting the tumor’s Reed-Sternberg cells from cytotoxic T cells involved in antitumor immune surveillance.
In contrast with the abundance of Tregs, Th1 CD4+ T cells and CD8+ cytotoxic T cells are rare in Hodgkin lymphoma biopsies and are not detected in the immediate proximity of Reed-Sternberg cells. The recruitment of cells involved in immune tolerance and the exclusion of lymphoid cells responsible for antitumor immune responses are explained by the expression of Reed-Sternberg cells of pro-Th2–associated cytokines, such as IL-4 and IL-13, and anti-Th1 or CD8 cytokines, such as IL-10 and transforming growth factor (TGF) β. The secretion of these immunosuppressive cytokines creates an immune-privileged microenvironment, allowing the tumor cells to avoid immune surveillance and T-cell–mediated apoptosis.
One of the most prominent features of Reed-Sternberg cells is the expression of CD30. Although coexpression of CD30 and its respective receptor CD153 initially suggested an autocrine mechanism promoting the proliferation of Reed-Sternberg cells via NF-κB, it is now well established that the activation of CD30 is primarily a CD153-independent process. Two additional members of the TNF receptor family, CD40 and receptor activator of nuclear factor κB (RANK), are expressed in Reed-Sternberg cells. Activation of CD40 seems to be mediated by the expression of CD40 ligand (CD40L) on T lymphocytes surrounding the Reed-Sternberg cell. Importantly, soluble CD40L induces proliferation and blocks CD95-induced apoptosis in Reed-Sternberg cells. Other factors that help rescue Hodgkin and Reed-Sternberg cells from an immunologic attack include the expression of the PD1 ligand and secretion of IL-10, TGF-β, and galectin 1 by the Reed-Sternberg cell, as well as inhibition of cytotoxic T cells by regulatory T cells (Tregs). Activation of RANK and osteoprotegerin, a member of the TNF-receptor superfamily, is triggered by the autocrine expression of RANK ligand (RANKL) in Reed-Sternberg cells. Activation of RANK promotes NF-κB activation and contributes to the maintenance of an inflammatory microenvironment by promoting interferon gamma (IFN-γ) and IL-13 secretion. Overall, the activation of TNF receptors by different mechanisms seems to play an important role in promoting the survival of the Reed-Sternberg cell.
Epidemiology and Causative Factors
The epidemiology of Hodgkin lymphoma is complex, with variation across geographic regions and with age, sex, and socioeconomic status. Data from cancer incidence surveys conducted in five continents have suggested that Hodgkin lymphoma has a bimodal peak at ages 15 to 34 years and a second peak in those older than 60 years. In Asian populations the overall incidence is only half that of the incidence in Europe. In all regions, Hodgkin lymphoma occurs rarely in children younger than 5 years of age ( Fig. 53-2 ).
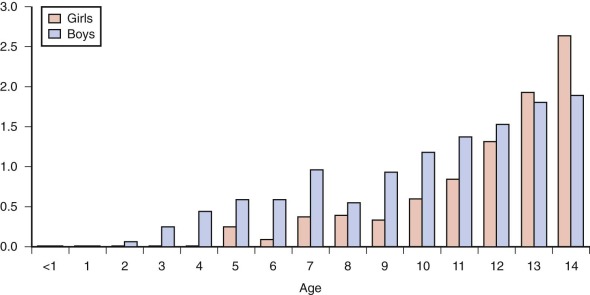
Hodgkin lymphoma occurring in patients younger than 15 years of age, referred to as childhood Hodgkin lymphoma, has several unique features compared with the more common disease of older adolescents and young adults. In childhood disease there is a 2 to 3 : 1 male preponderance, whereas in older patients the disease is almost equal in males and females. Within the younger age group, mixed cellularity and nodular lymphocyte-predominant histologies are more common. In addition, in childhood Hodgkin lymphoma increasing family size and lower socioeconomic status are risk factors for disease. Conversely, in Hodgkin lymphoma in adolescents and young adults, higher socioeconomic status and smaller family size are risk factors for disease.
The role of EBV as an etiologic agent for Hodgkin lymphoma has been the source of much study. In 1966 MacMahon was the first to suggest an infectious cause of Hodgkin lymphoma, and in 1974 Rosdahl and colleagues reported an increased risk of Hodgkin lymphoma in people with a history of infectious mononucleosis. Molecular studies identified monoclonal EBV genomes in Reed-Sternberg cells, implying that they were infected before malignant transformation. Subsequent studies showed EBV genome sequences associated with Reed-Sternberg cells in 59% of cases. The proportion of EBV-positive Hodgkin lymphoma cases varies, however, according to geographic region, age, and histologic subtype. The proportion of EBV-positive Hodgkin lymphoma cases is higher in children than adults. It is most prevalent in mixed cellularity histology and more common in boys. The incidence is higher in the developing world including parts of Africa, Asia, and South America, with an incidence of EBV-positive Hodgkin lymphoma as high as 90% in Peru.
Familial aggregation of Hodgkin lymphoma was first described by Razis and coworkers in 1959 and is now well documented. The familial risk of Hodgkin lymphoma ranks among the highest in the population-based Swedish Family Cancer Database. In an analysis of 28 reports of familial Hodgkin lymphoma there is only one major peak between 15 and 34 years of age for familial Hodgkin lymphoma instead of the classic bimodal age distribution of sporadic Hodgkin lymphoma. This corresponds to the findings of two large studies of an increased sevenfold risk in siblings of cases diagnosed at ages younger than 45 and 35 years of age, respectively, but little or no increased risk in siblings of cases diagnosed at older ages. Strong evidence for a role for genetic susceptibility was provided by the finding that monozygotic twins of patients with Hodgkin lymphoma have a 99-fold increased risk, whereas no increased risk in dizygotic twins was observed.
Familial aggregation of Hodgkin lymphoma may in part reflect inherited abnormalities of the immune response. A familial or personal history of autoimmune conditions and sarcoidosis is associated with an increased risk for Hodgkin lymphoma. A 51-fold increased risk for Hodgkin lymphoma was found in kindreds predisposed to autoimmune lymphoproliferative syndrome, a disorder of lymphocyte homeostasis usually associated with germline FAS gene mutations. In patients with ataxia-telangiectasia mutation (ATM), the risk of Hodgkin lymphoma was increased, but its risk was much lower than the risk of NHL. In 1975 Svejgaard and coworkers first described an association of certain human leukocyte antigen (HLA) loci with an increased risk of Hodgkin lymphoma, suggesting a disease susceptibility gene within or near the histocompatibility region on chromosome 6. Since then, numerous studies have confirmed the association of particular HLA loci in the susceptibility to Hodgkin lymphoma, including HLAs A1, B5, B8, B15, B27, B35, and B37. In a large study on familial Hodgkin lymphoma, Chakravarti and colleagues have found strong evidence for a recessive mode of inheritance for susceptibility to Hodgkin lymphoma, and approximately 60% of associations were caused by an HLA-linked susceptibility gene. Recent genetic studies have identified disruption of KLHDC8B in the germline of a family with several cases of Hodgkin lymphoma and a germline frameshift mutation in the NPAT gene in a family with four cases of NLPHL. Finally a genome-wide association study has identified risk loci at 2p16.1 ( REL ), 8q24.21 ( PVT1 ), and 10p14 ( GATA3 ) and confirmed the strong HLA association with this disease.
Clinical Characteristics
The most common clinical presentation of Hodgkin lymphoma in children and adolescents is a persistently enlarged node in the cervical or supraclavicular region. Characteristically the lymph nodes involved with Hodgkin lymphoma are not painful and have a “rubbery” firmness on palpation. Enlarged nodes have often been present for weeks or months, increasing and decreasing in size irrespective of whether antibiotic therapy has been given. Although the majority of patients have painless cervical adenopathy, the clinical presentation varies considerably, ranging from life-threatening airway compression to the coincidental detection of an enlarged node during an otherwise routine examination. Approximately 80% of children have disease in one or both sides of the upper or lower neck. Of those with cervical adenopathy, more than two thirds have intrathoracic disease, most commonly in the anterosuperior mediastinum, paratracheal, and tracheobronchial lymph node groups. Pulmonary parenchymal involvement is rarely observed in the absence of hilar disease. Pleural effusions are uncommon; they are usually an indication of lymphatic obstruction from bulky central disease rather than a sign of advanced-stage disease. Pericardial effusion may occur in cases with pericardial involvement and occurs most often in the setting of bulky mediastinal disease. Hodgkin lymphoma has a strong tendency for contiguous spread along adjacent lymph-node regions. Although about 30% of patients have supradiaphragmatic and infradiaphragmatic disease, Hodgkin lymphoma limited to infradiaphragmatic sites is rare.
Systemic symptoms including fatigue and anorexia are common in patients with Hodgkin lymphoma. At the time of diagnosis, approximately 30% of patients have constitutional signs referred to as B symptoms and defined as the presence of fever (temperature higher than 38° C [100.4° F]) for 3 consecutive days, drenching night sweats and unexplained body weight loss of 10% or more over the preceding 6 months. Classically the fever associated with Hodgkin lymphoma, called the Pel-Epstein fever, occurs in the evening and becomes more pronounced with time. Severe and unexplained pruritus is associated with Hodgkin lymphoma, sometimes preceding the development of adenopathy. A poorly understood and relatively unusual sign of Hodgkin lymphoma is pain at the sites of disease with alcohol ingestion.
Laboratory findings are often nonspecific with mild anemia and elevated erythrocyte sedimentation rate (ESR) being the most common findings. Nonspecific hematologic laboratory findings include neutrophilia, monocytosis, lymphopenia, and eosinophilia. Acute-phase reactants including C-reactive protein, ferritin, and serum copper may be elevated. High levels of alkaline phosphatase can be associated with bone involvement.
Rarely patients with Hodgkin have findings suggestive of paraneoplastic syndromes such as nephritic syndrome, polymyositis, idiopathic cholestasis and autoimmune hemolytic anemia, neutropenia, and thrombocytopenia, or combinations of these. Limbic encephalitis or subacute cerebellar degeneration have also been reported as very rare paraneoplastic syndromes in patients with Hodgkin lymphoma.
At the time of diagnosis patients may exhibit altered immune function characterized by reduced cellular immunity, whereas humoral immunity is usually relatively intact. The nature of the immune defect is unclear. The severity of the impairment increases with advanced stage, disease progression, and recurrence and after treatment with radiotherapy and chemotherapy. T-cell deficits may persist for a prolonged period in successfully treated patients.
Diagnosis and Staging
Biopsy to provide pathologic tissue for examination is required for the diagnosis of Hodgkin lymphoma. Excisional biopsy of an enlarged node is preferred. Samples from needle biopsies are often insufficient for the diagnosis of Hodgkin lymphoma because of the importance of the information provided by the nodal architecture and background stroma, as well as the need to identify the relatively rare Reed-Sternberg cell. In a case series, one third of children who ultimately were diagnosed with Hodgkin lymphoma and who underwent core needle biopsies of their mediastinal masses were not able to be diagnosed from these initial samples. Needle biopsy should be restricted to situations in which surgery and general anesthesia may carry undue risks for the patient. If needle biopsy is performed, multiple biopsy samples should be taken to augment diagnostic potential.
The purpose of a staging evaluation is to identify all sites and characteristics of Hodgkin lymphoma in each patient to permit accurate stratification of therapy based on risk, the definition of areas to be included in potential radiation fields, and to inform follow-up imaging studies. Table 53-2 gives an overview of suggested evaluations.
| Laboratory studies |
|
| Imaging |
|
| Other investigations |
|
| Other pretherapy considerations |
|
A plain film of the chest is useful in providing preliminary information about mediastinal involvement and is an essential evaluation in a patient who will undergo anesthesia for cervical node biopsy ( Fig. 53-3 ). Computed tomography (CT) of the neck, chest, abdomen, and pelvis are standard initial staging evaluations. In the past, imaging of the neck was considered not necessary given the ability to detect cervical node by physical examination. Imaging of the neck, however, can be of significant importance to current care, because it allows for the most accurate radiation field planning, avoiding potential overtreatment of the neck, and providing baseline data for the measurement of disease response. CT of the chest provides detailed information about sites of disease involvement including the mediastinum, pulmonary parenchyma, pleura, and pericardium. Sites of disease in the abdomen and pelvis are visualized with CT done with oral and intravenous (IV) contrast. Alternatively, magnetic resonance imaging (MRI) or ultrasound can also be used for staging abdominal and pelvis disease.

Functional nuclear imaging is an important component to initial staging though perhaps has an even more significant role in assessment of disease response (see later). Gallium-67 imaging has largely been replaced by 18-fluorodeoxyglucose positron emission tomography (FDG-PET) because of increased sensitivity and specificity. Increased F-FDG uptake in lymphoma is based on elevated glycolysis and the longer residence time of 18 F-FDG in malignant cells compared with most normal tissues. Numerous studies in adults with Hodgkin lymphoma have demonstrated that PET is able to detect an additional number of Hodgkin lymphoma lesions compared with conventional imaging studies, in particular CT and bone marrow biopsy, resulting in a modification of staging in 15% to 20% of patients, with an impact on disease management in 5% to 15% of cases. The data in children are more limited. A review of a single-center experience showed that the findings from the diagnostic FDG-PET were more sensitive than conventional imaging and that the findings altered the involved-field radiation treatment fields in 17% of patients. Prospective studies are required to assess whether FDG-PET done at the time of diagnosis has on impact on outcome or on treatment burden.
Historically, surgical staging including splenectomy was considered standard for patients with Hodgkin lymphoma. Staging splenectomy is associated with significant risks. In the German-Austrian experience of 1181 children with Hodgkin lymphoma, the survival was poorer for children younger than age 10 years because of episodes of infection and death caused by sepsis in splenectomized children. With the availability of cross-sectional imaging techniques, routine staging laparotomy has now been abandoned. In the relatively rare circumstance of imaging findings that are ambiguous in defining disease involvement or noninvolvement, and when this information affects therapeutic decision making and in particular radiation fields, then staging biopsy may still be warranted.
Until recently bone marrow biopsy has been considered a standard component of the initial staging evaluation for any child with cytopenias at presentation and all patients except for those with stage 1A or 2A disease. Retrospective investigations have suggested that the impact of the findings from diagnostic bone marrow biopsies had a minimal effect on patient-risk assignment and subsequent therapy. FDG-PET is a sensitive and specific method for detection of bone marrow disease. Two recent studies provide data to support the idea that FDG-PET can replace bone marrow biopsies for staging purposes in an analogous fashion to cross-sectional imaging of the abdomen replacing staging laparotomy in the past.
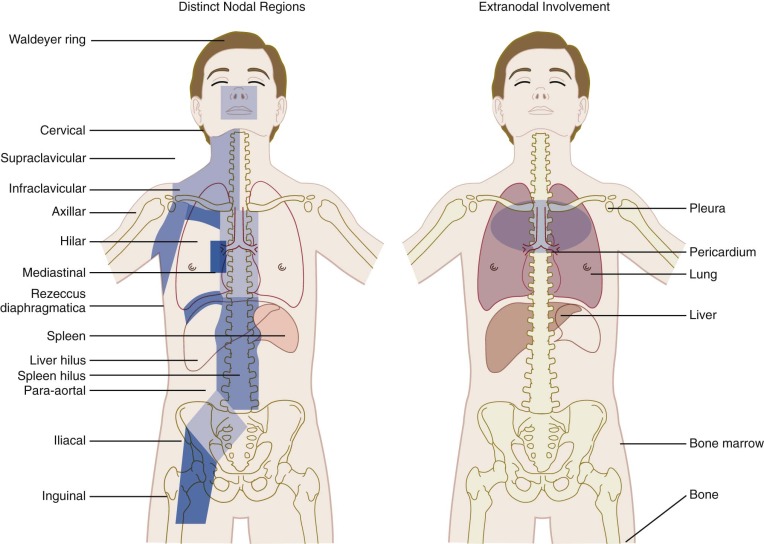
The Ann Arbor staging classification for Hodgkin lymphoma, adopted in 1971, was based on the recognized orderly spread of the disease between contiguous lymph nodes, which predominates until late in the course of the disease ( Table 53-3 ). The distinct lymph node regions recognized by the Ann Arbor classification system are shown in Figure 53-4 . The substage classifications A, B, and E amend each stage based on distinct features. Stage A designates asymptomatic disease; B indicates the presence of any one of the three B symptoms as defined in Table 53-3 ; and extralymphatic disease is designated as E, referring to limited extranodal extensions that easily can be encompassed within a radiotherapy portal. Extralymphatic disease is further designated as L (lung), P (pleura), and O (osseous) according to this system. The decision to classify extralymphatic disease either as substage E or as stage IV is based on the clinician’s judgment and often depends on whether the extralymphatic disease can be adequately covered in a radiotherapy treatment portal. Multiple E lesions are automatically considered to be stage IV.
| Stage | Description |
|---|---|
| I | Involvement of a single lymph-node region (I) or a single extralymphatic organ or site (I E ) |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm (II) or localized contiguous involvement of only one extralymphatic organ or site and its regional lymph node(s) on the same side of the diaphragm (II E ) |
| III | Involvement of lymph-node regions on both sides of the diaphragm (III), which may also be accompanied by involvement of the spleen (III S ) or by localized contiguous involvement of an extralymphatic organ or site (III E ) or both (III SE ) |
| IV | Diffuse or disseminated involvement of one or more extralymphatic organs or tissues, with or without associated lymph-node involvement |
| Designations Applicable to Any Stage | |
| A | No B symptoms |
| B | B symptoms, defined as presence of fever (>38° C [100.4° F]) for 3 consecutive days, drenching night sweats, or unexplained loss of 10% or more of body weight in the preceding 6 months |
| E | Involvement of a single extranodal site that is contiguous or proximal to the known nodal site |
Treatment
Centers treating children with Hodgkin lymphoma should have extensive experience and a dedicated multidisciplinary team, including a pediatric surgeon, radiation oncologist, pediatric oncologist, pathologist, and diagnostic radiologist. If such a team is not available at the facility at which the child is initially seen, prompt referral to a comprehensive childhood cancer center is essential.
Prognostic Factors/Stratification of Treatment.
As with other malignancies, prognostic factors are useful as tools for defining risk groups for stratification of treatment intensity. For patients with Hodgkin lymphoma this translates into both the choice of chemotherapeutic regimen and the use of radiotherapy. The stage of disease is a strong prognostic factor and widely used for risk stratification that has been used in all major cooperative groups. Apart from the stage of disease, a number of factors have been reported to be associated with the risk of treatment failure, including the presence of B symptoms, histologic subtype, bulky disease, gender, anemia, ESR, and evidence of latent EBV infection, some of which have lost significance with improvements in therapy. The prognostic value of these variables has not been consistent across studies, in part related to treatment prescribed. For example, mediastinal bulky disease—defined as a mass larger than one third of the maximum chest diameter—was associated with increased risk for relapse in some studies but not in the German-Austrian trial DAL-HD90. However, in this protocol patients with a larger residual mass at the end of chemotherapy received a boost dose in addition to the involved field irradiation. Similarly, male gender had an adverse impact on outcome in that trial. For boys, however, procarbazine was partially replaced by etoposide, and girls received a full dose of procarbazine. Race and ethnicity have been identified as risk factors in some pediatric cancers. In a single-center retrospective review, African-American children had an inferior event-free survival (EFS) rate but an identical overall survival (OS) as compared with white children.
The risk-group definitions used in selected clinical trials are described in Table 53-4 . In general in addition to stage, disease bulk and presence of B symptoms are the factors used. The kinetics of disease response to chemotherapy, measured by CT and/or PET scan is used in many cooperative group trials as an important additional tool for the stratification of treatment intensity including the use of radiation (see below).
| Study Group | Stage | ||
|---|---|---|---|
| Low Risk | Intermediate Risk | High Risk | |
| DAL-HD90 | IA, IB, IE, IIA | IIEA, IIB, IIIA | IIEB, IIIEA, IIIB, IIIEB, IV |
| GPOH-HD2002 | IA, IB, IIA | IE, IIEA, IIB, IIIA | IIEB, IIIEA, IIIB, IV |
| CCG 5942 | I + IIA, without * | I + II with * IIB, III | IV |
| Stanford, St. Jude, Boston Consortium | I, II, without † | “Unfavorable” I, II with † III, IV | |
| POG | I, IIA, IIIA1 | “Unfavorable” IIB, IIIA 2 , IIIB, IV | |
| COG | IA, IIA without ‡ | IA+IIA with bulk, IB+IIB, IIIA, IVA | IIIB + IVB |
* Adverse disease features (one or more of the following): hilar adenopathy, more than four nodal regions, mediastinal tumor >33% of chest diameter, node or nodal aggregate with diameter >10 cm.
† Presence of one or more of the following: bulky disease, peripheral nodal disease 6 cm or larger, and/or mediastinal tumor one third of intrathoracic diameter or more, and B symptoms.
Treatment and Outcomes.
In the 1960s extended-field radiation with doses of 35 to 44 Gy were shown to be curative in a significant portion of patients with Hodgkin lymphoma. In children, the negative sequelae of high-dose extended-field radiation quickly became apparent. Soon thereafter the MOPP combination was created by Devita and coworkers to treat patients with Hodgkin lymphoma. The basic rationale was to combine single drugs with proven efficacy but different mechanisms of action and resistance that had few overlapping toxicities to maximize antitumor effect and limit side effects caused by use of moderate doses of the individual drugs. The second important combination chemotherapy regimen for the treatment of Hodgkin lymphoma was doxorubicin (Adriamycin), bleomycin, vinblastine, and dacarbazine (ABVD), which was developed by Bandanna and coworkers. Variations of these two regimens have formed the cornerstone of almost all subsequent chemotherapy regimens.
Combined-modality therapy using both chemotherapy and radiation has emerged as the standard of care for most children and adolescents diagnosed with Hodgkin lymphoma. The use of six cycles of MOPP chemotherapy combined with involved-field radiotherapy (IFRT) was pioneered by the Stanford investigators in children with pathologically staged disease. This pivotal trial has served as the model for combined-modality treatment programs in children.
Subsequent studies evaluated regimens with modifications incorporated designed to minimize risks of late effects while maintaining efficacy. Changes have included both the substitution of drugs the use of which is associated with lower risks of specific long term toxicities as well as limiting the cumulative dose of drugs that are associated with risks of late sequelae in a dose dependent manner. Some modifications have been gender specific. Table 53-5 gives an overview of chemotherapy combinations. With most of these multiagent combinations, comparable high disease-free survival rates have been achieved.
| Course | Drugs | Dosage and Route | Days |
|---|---|---|---|
| MOPP | Mechlorethamine | 6.0 mg/m 2 , IV | 1, 8 |
| Vincristine (Oncovin) | 1.4 mg/m 2 , IV | 1, 8 | |
| Procarbazine | 100 mg/m 2 , PO | 1-15 | |
| Prednisone | 40 mg/m 2 , PO | 1-15 | |
| MOPP Derivatives | |||
| COPP | Cyclophosphamide | 600 (500) mg/m 2 , IV | 1, 8 |
| Vincristine (Oncovin) | 1.4 mg/m 2 , IV (max 2 mg) | 1, 8 | |
| Procarbazine | 100 mg/m 2 , PO | 1-15 | |
| Prednisone | 40 mg/m 2 , PO | 1-15 | |
| COMP | Cyclophosphamide | 600 mg/m 2 , IV | 1, 8 |
| Vincristine (Oncovin) | 1.4 mg/m 2 , IV (max 2 mg) | 1, 8 | |
| Methotrexate | 40 mg/m 2 , IV | 1, 8 | |
| Prednisone | 40 mg/m 2 , PO | 1-15 | |
| COP | Cyclophosphamide | 600 mg/m 2 , IV | 1, 8 |
| Vincristine (Oncovin) | 1.4 mg/m 2 , IV (max 2 mg) | 1, 8 | |
| Procarbazine | 100 mg/m 2 , PO | 1-14 | |
| ABVD | Doxorubicin (Adriamycin) | 25 mg/m 2 , IV | 1, 15 |
| Bleomycin | 10 U/m 2 , IV | 1, 15 | |
| Vinblastine | 6 mg/m 2 , IV | 1, 15 | |
| Dacarbazine | 375 mg/m 2 , IV | 1, 15 | |
| MOPP-ABVD Hybrids and Derivatives | |||
| OPPA | Vincristine (Oncovin) | 1.5 mg/m 2 , IV (max 2 mg) | 1, 8, 15 |
| Procarbazine | 100 mg/m 2 , PO | 1-15 | |
| Prednisone | 60 mg/m 2 , PO | 1-15 | |
| Doxorubicin (Adriamycin) | 40 mg/m 2 , IV | 1, 15 | |
| OPA | Vincristine (Oncovin) | 1.5 mg/m 2 , IV (max 2 mg) | 1, 8, 15 |
| Prednisone | 60 mg/m 2 , PO | 1-15 | |
| Doxorubicin (Adriamycin) | 40 mg/m 2 , IV | 1, 15 | |
| OEPA | Vincristine (Oncovin) | 1.5 mg/m 2 , IV (max 2 mg) | 1, 8, 15 |
| Etoposide | 125 mg/m 2 , IV | 3-6 | |
| Prednisone | 60 mg/m 2 , PO | 1-15 | |
| Doxorubicin (Adriamycin) | 40 mg/m 2 , IV | 1, 15 | |
| ChlVPP | Chlorambucil | 6 mg/m 2 , PO | 1-14 |
| Vinblastine | 6 mg/m 2 , PO | 1, 8 | |
| Procarbazine | 100 mg/m 2 , PO | 1-14 | |
| Prednisone | 40 mg/m 2 , PO | 1-14 | |
| VAMP | Vinblastine | 6 mg/m 2 , IV | 1, 15 |
| Doxorubicin (Adriamycin) | 25 mg/m 2 , IV | 1, 15 | |
| Methotrexate | 20 mg/m 2 , IV | 1, 15 | |
| Prednisone | 40 mg/m 2 , PO | 1-14 | |
| VBVP | Vinblastine | 6 mg/m 2 , IV | 1, 8 |
| Bleomycin | 10 mg/m 2 , IV | 1 | |
| Etoposide (VP-16) | 100 mg/m 2 , IV | 1-5 | |
| Prednisolone | 40 mg/m 2 , PO | 1-8 | |
| COPP-ABV | Cyclophosphamide | 600 mg/m 2 , IV | 0 |
| Vincristine (Oncovin) | 1.4 mg/m 2 , IV | 0 | |
| Procarbazine | 100 mg/m 2 , PO | 0-6 | |
| Prednisolone | 40 mg/m 2 , PO | 0-13 | |
| Doxorubicin (Adriamycin) | 35 mg/m 2 , IV | 7 | |
| Bleomycin | 10 U/m 2 , IV | 7 | |
| Vinblastine | 6 mg/m 2 , IV | 7 | |
| ABC | A | ||
| Cytarabine | 3 g/m 2 , IV over 3 hr, q12h | 0, 1 | |
| Etoposide | 200 mg/m 2 , IV over 1 hr, q12h | 0, 1 | |
| G-CSF | 5 µg/kg, SC | Starting on day 2 | |
| B | |||
| COPP-ABVD | See COPP/ABVD | 21-27 | |
| G-CSF | 5 µg/kg, SC | Starting on day 28 | |
| C | |||
| Vincristine | 1.4 mg/m 2 , IV | 42 | |
| Cyclophosphamide | 1200 mg/m 2 , IV | 42-43 | |
| Doxorubicin | 25 mg/m 2 /day, continuous infusion | 42-44 | |
| Methylprednisolone | 250 mg/m 2 , IV q6h | 42 | |
| Prednisone | 60 mg/m 2 , PO | 43-46 | |
| G-CSF | 5 µg/kg, SC | Starting on day 46 | |
| BEACOPP | Bleomycin | 10 U/m 2 , IV | 8 |
| Etoposide | 200 mg/m 2 , IV | 1-3 | |
| Doxorubicin (Adriamycin) | 35 mg/m 2 , IV | 1 | |
| Cyclophosphamide | 1200 mg/m 2 , IV | 1 | |
| Vincristine (Oncovin) | 2 mg/m 2 (max 2 mg), IV | 8 | |
| Procarbazine | 100 mg/m 2 , PO | 1-7 | |
| Prednisone | 40 mg/m 2 , PO | 1-14 | |
| DBVE | Doxorubicin | 25 mg/m 2 , IV | 1, 15 |
| Bleomycin | 10 mg/m 2 , SC | 1, 15 | |
| Vincristine | 1.5 mg/m 2 (max,] 2 mg), IV | 1, 15 | |
| Etoposide | 100 mg/m 2 , IV over 1 hr | 1-5 | |
| Stanford V | Mechlorethamine | 6 mg/m 2 , IV | 1 |
| Vinblastine | 6 mg/m 2 , IV | 1, 15 | |
| Doxorubicin | 25 mg/m 2 , IV | 1, 15 | |
| Etoposide | 60 mg/m 2 , IV | 15, 16 | |
| Vincristine | 1.4 mg/m 2 (max 2 mg), IV | 8, 22 | |
| Bleomycin | 5 U/m 2 , IV | 8, 22 | |
| Prednisone | 40 mg/m 2 , PO | Every other day | |
| ABVD-PC | Doxorubicin (Adriamycin) | 25 mg/m 2 IV | 1, 2 |
| Bleomycin | 10 IU/m 2 * IV | 1, 8 | |
| Vincristine | 1.4 mg/m 2 IV (max 2.8) | 1, 8 | |
| Etoposide | 125 mg/m 2 IV | 1-5 | |
| Prednisone | 40 mg/m 2 , PO | 1-7 | |
| Cyclophosphamide | 800 mg/m 2 IV | 1 | |
| G-CSF | 5 µg/kg SC | Starting day 6 | |
The balance of efficacy and late risk of the combined-modality of chemotherapy and radiotherapy adopted by the German-Austrian study group has been investigated over a series of trials ( Table 53-6 ). Modifications investigated have included reduction of the cumulative dose of alkylating agents and replacement of mechlorethamine of MOPP with doxorubicin (vincristine [Oncovin], prednisone, procarbazine, and doxorubicin [OPPA]) and cyclophosphamide (cyclophosphamide, vincristine [Oncovin], procarbazine, and prednisone [COPP]); reduction of radiotherapy (volume, dose, and number of patients who receive radiotherapy); and replacement of procarbazine by etoposide for boys. The seventh of the sequential studies built on findings of the previous six trials. In this study the primary aim was to evaluate modifications of therapy to maintain good outcomes for boys but to do so with less gonadotoxic therapy. In this study procarbazine was replaced with higher dose etoposide in the first two cycles and with dacarbazine in the subsequent two to four cycles. The outcomes for boys and girls on this study were superimposable, supporting the efficacy of this regimen.
| Study | No. of Patients (EFS) | Therapy Group 1 | Therapy Group 2 | Therapy Group 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Stages (% of patients) EFS | Chemotherapy | Radiotherapy | Stages (% of patients) EFS | Chemotherapy | Radiotherapy | Stages (% of patients) 5-yr EFS | Chemotherapy | Radiotherapy | ||
| DAL-HD78 | 170 (92%) | I, IIA (43%) 95% | OPPA ×2 | EF, 36-40 Gy | >IIA (57%) 89% | OPPA ×2 COPP ×4 | EF, 36-40 Gy | Included in TG2 | ||
| DAL-HD82 | 203 (96%) | I, IIA (49%) 99% | OPPA ×2 | IF, 35 Gy | IIB, IIIA (26%) 96% | OPPA ×2 COPP ×2 | IF, 30 Gy * | IIIB, IV (25%) 87% | OPPA ×2 COPP ×4 | 25 Gy * |
| DAL-HD85 | 103 (77%) | I, IIA (58%) 59% | OPA ×2 | IF, 35 Gy | IIB, IIIA (20%) 62% | OPPA ×2 COMP ×2 | IF, 30 Gy * | IIIB, IV (22%) 62% | OPA ×2 COMP ×4 | 25 Gy * |
| DAL-HD87 | 204 (85%) | I, IIA (51%) 84% | OPA ×2 | IF, 30 Gy * | IE, IIEA IIB, IIIA (17%) 82% | OPPA ×2 COPP ×2 | 25 Gy * | IIIB, IV (28%) 89% | OPPA ×2 COPP ×4 | 25 Gy * |
| DAL-HD90 | 578 (91%) | I, IIA (46%) 94% | OPPA ×2 † OEPA ×2 ‡ | IF, 25 Gy * | IE, IIEA IIB, IIIA (21%) 93% | OPPA ×2 † COPP ×2 OEPA ×2 ‡ COPP ×2 | 25 Gy * | IIEB, IIIEA IIIB, IV (31%) 86% | OPPA ×2 † COPP ×4 OEPA ×2 ‡ COPP ×4 | 20 Gy * |
| GPOH-HD95 | 1018 (88%) | I, IIA (40%) 94% | OPPA ×2 † OEPA ×2 ‡ | CR: no RT (28%) § ; non-CR: IF, 20 Gy * | IE, IIEA IIB, IIIA (26%), 87% | OPPA ×2 † COPP ×2 OEPA ×2 ‡ COPP ×2 | CR: no RT (19%) § ; non-CR: IF 20 Gy * | IIEB, IIIEA; IIIB, IV (33%) 83% | OPPA ×2 † COPP ×4 OEPA ×2 ‡ COPP ×4 | CR: no RT (17%) § ; non-CR: IF, 20 Gy * |
| GPOH-HD02 | 573 (89%) | 1A/B, IIA (34%) 92% | OPPA ×2 † OE*PA ×2 ‡ | CR: no RT (33%) § ; non-CR: IF, 20 Gy * | IE, IIEA, IIB, IIIA (24%) 93% | OPPA ×2 † COPP ×2 OEPA ×2 ‡ COPDAC ×2 | 20 Gy * | IIEB, IIIEA; IIIB, IV (42%) 87% | OPPA, ×2 † COPP ×4 OEPA ×2 ‡ COPDAC ×4 | 20 Gy * |
* Boost to residual tumor to 30-35 Gy.
§ Percentage of patients in the treatment subgroup who did not receive radiotherapy.
The second strategy that is of critical importance in trying to maximize efficacy and minimize toxicity is the stratification of treatment intensity based on the patient’s risk for relapse. Cooperative group trials of pediatric and adolescent Hodgkin lymphoma have generally divided patients into low-, intermediate-, and high-risk groups (see Table 53-4 ). In addition to stratification by stage, studies have also evaluated modifications of therapy based on early disease response. Many reports from these therapeutic studies are confined to a distinct risk group of patients. Comparisons among trials confined to patient subsets need to be done with caution, because the risk group definitions vary both based on the patient characteristics at the time of initial staging and the criteria of disease response.
Table 53-7 summarizes treatment strategy and results of therapeutic studies in the patient subgroup defined as low risk. Patients at low risk in general are those with stage I and II disease without B symptoms and without bulky disease. These patients have been shown to have excellent outcomes with sequential trials using two to four cycles of multiagent chemotherapy and low-dose involved-field radiation. These regimens have been designed to minimize exposure to anthracyclines, alkylators, and epipodophyllotoxins in order to minimize risks of cardiac dysfunction, infertility, and secondary acute myelogenous leukemia (AML), respectively.
| Group or Institution | No. of Patients | Stage | Chemotherapy * | Radiation (Gy), Field | EFS or RFS Overall (%) | Survival (%) |
|---|---|---|---|---|---|---|
| GPOH-HD95 | 281 | I, IIA | Female: OPPA ×2 Male: OEPA ×2 | CR † : no RT (22%); non-CR: IF, 20 Gy; residual tumor, 30-35 Gy | 94 | NR |
| GPOH-HD02 | 195 | IA/B, IIA | Female: OPPA ×2 Male: OE*PA ‡ ×2 | CR † : no RT (33%); no-CR: IF, 20 Gy; residual tumor, 30-35 Gy | 92 | 99 |
| French Society of Pediatric Oncology | 202 | I, II I, II | 4 VBVP, good responders; 4 VBVP + 1 or 2 OPPA, poor responders | 20, IF 20, IF | 91 and 78 § | 97.5 |
| Stanford-St. Jude-Boston Consortium | 88 | 1, II ‖ | 4 VAMP | CR post 2 cycles, no IF No CR: 25.5 Gy | 90 (2 yr) | 99 |
| CCG5942 | 294 | I + IIA without ¶ | COPP-ABV × 4 | CR, ** random: IF 21 Gy vs. no RT; PR, 21 Gy IF | 95 (3 yr) | 100 |
| POG9226 | 51 | I, IIA, IIIA1 | 4 DBVE | 25, IF | 91(6 yr) | 98 |
* For the composition of combination chemotherapy courses, see Table 53-8 .
† CR defined as reduction of tumor volume of >95% and residual mass <2 mL.
‡ OE*PA includes higher dose of etoposide than OEPA.
§ For good and poor responders respectively.
‖ Less than 3 nodal sites, no B symptoms, no mediastinal bulk, no extranodal extension.
¶ Peripheral nodal disease, <6 cm, mediastinal tumor smaller than one third of intrathoracic diameter, absence of extranodal involvement.
** CR defined as >70% reduction of initial tumor volume and gallium-negative if initially gallium-positive.
Table 53-8 summarizes the treatment strategy and results of selected therapeutic studies in patients considered to have intermediate- or high-risk disease. Chemotherapy for these groups generally consists of combinations of ABVD and MOPP or hybrid regimens. Most regimens contain four to six cycles of chemotherapy followed by involved-field radiation with some including high-dose radiation to bulky or residual sites of disease.
| Group or Institution | No. of Patients | Stage | Chemotherapy | Radiation (Gy), Field | EFS at 5 years | Overall Survival (%) |
|---|---|---|---|---|---|---|
| DAL-HD90 (high risk) | 179 | IIEB, IIIEA, B, IIIB, IVA, B | 2 OEPA-OPPA + 4 COPP | IF, 20-25 Gy residual tumor 30-35 Gy | 86% | 94% |
| GPOH-HD95 3-5 ABVD-PC | 265 | IIEA, IIIEA, B, IIIB, IVA, B | 2 OPPA/OEPA + 4 COPP | CR * : no RT (22%); non-CR: IF, 20 Gy; residual tumor, 30-35 | 79% DFS no IFRT 91% DFS with IFRT | NR |
| GPOH-HD02 (intermediate risk) | 139 | IAE, IB, IIAE, IIB, IIIA | 2 OPPA + 2 COPP † 2 OE*PA +2 COPAD ‡ | IF, 20 Gy; residual tumor, 30-35 | 93% | 98% |
| GPOH-HD02 (highest risk) | 239 | IIBE, IIIAE, IIIB, IVA, IVB, IVE | 2 OPPA + 4 COPP † 2 OE*PA + 4 COPAD ‡ | IF, 20 Gy; residual tumor, 30-35 | 87% EFS | 95% 5 yrs |
| Stanford-St. Jude-Boston Consortium | 159 | I-II unfavorable, § III, IV | 3 VAMP/3 COP | 15-25.5 Gy, ‖ IF | 76 at 5 yr | 93% at 5 yr |
| POG | 179 | IIB, IIIA2, IIIB, IV | 4 MOPP, 4 ABVD | 21 Gy total nodal | 79% | 92% |
| POG | 216 | IB, IIA/IIIA with bulk, IIB, IIIB, IV | 3-5 ABVD-PC | 21 Gy IF | 84% at 5 yr | 95% at 5 yr |
| CCG | 394 | I/IIB, IIB, III | 6 COPP-ABV | CR: randomized to 21 Gy vs. no IFRT, no CR 21 Gy IFRT | 87% | 95% |
| CCG | 141 | IV | COPP-ABV + CHOP + Ara-C | CR: randomized to 21 Gy vs. no IFRT, no CR 21 Gy IFRT | 90% | 100% |
| COG | 98 | IIB with bulk, ¶ IIIB with bulk, IV | BEACOPP × 4 then one of: COPP/ABV × 4 ‡ ABVD × 2 † BEACOPP × 4 # | Female, RER ** : no RT Male or SER: 21 Gy Residual tumor, 35 Gy | 94% EFS | 97% 5 yr |
* CR defined as reduction of tumor volume of >95% and residual mass <2 mL.
§ Defined as presence of one or more of the following: bulky disease, peripheral nodal disease 6 cm or larger, and/or mediastinal tumor one third of intrathoracic diameter or larger; and B symptoms.
‖ 15 Gy for nonbulky sites and complete response after two cycles of chemotherapy.
¶ Bulk defined as mediastinal mass greater than intrathoracic diameter at level of T5 or extrathoracic nodal mass greater than 10cm.
** SER, partial response not meeting criteria for RER; RER, >70%reduction in tumor volume and gallium negative post 4 cycles.
Prognostic information for patients with Hodgkin lymphoma is based on information available at the time of the child’s initial evaluation. Additionally the importance of the kinetics of disease response to chemotherapy has been shown to be a predictive factor for this disease, as it has been for pediatric leukemias. The premise is that those with more robust responses to initial chemotherapy have more favorable disease and that responsiveness can be included in the factors that are used to decide the intensity of therapy, including whether irradiation is indicated. The definition of a favorable response to chemotherapy, including the timing of assessment of response, the intensity of the chemotherapy regimen used to get the response, and the tools used to measure response have varied widely between study groups and over time. Table 53-9 describes the criteria for disease response evaluations in recent cooperative group trials.
| Study | Definition of Favorable Response | Timing of Assessment | Treatment Modification for Those with Favorable Response |
|---|---|---|---|
| CCG5942 | >70% reduction in initial tumor volume and gallium negative | At completion of chemotherapy | Randomized to IFRT or no IFRT |
| GPOH-HD02 | ≥95% reduction in tumor volume | After 2, 4, or 6 cycles of chemotherapy depending on disease group | No IFRT for patients with low-stage disease only (TG1) |
| Stanford-St. Jude-Boston Consortium | ≥75% reduction in perpendicular diameters of measurable lesions, or return no normal size and gallium or PET negative | After 2 cycles of VAMP | No IFRT |
| COG | Disappearance of all clinical and disease-related symptoms and PET negative | After 2 cycles of ABVD-PC | All get IFRT; Less than good response: chemo intensified with additional 2 cycles ifos/vinolebine |
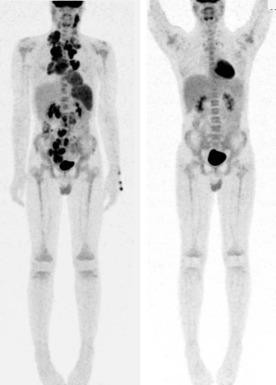
Cross-sectional imaging can define response based on a percentage decrease in size. In Hodgkin lymphoma, a significant proportion of patients have residual masses at the time of disease response assessment and at the end of therapy. Functional imaging methods such as gallium scanning and PET are able to assess metabolically active tissue associated with residual disease ( Fig. 53-5 ). A series of studies primarily performed in adults with Hodgkin lymphoma where therapy was not changed based on interim radiographic results have shown that PET scans performed after two cycles of chemotherapy are highly predictive of outcome. The accuracy of PET to predict treatment outcome is higher early in the course of treatment as compared with the end of chemotherapy. Both the German-Austrian study GPOH-HD2002 and the Children’s Oncology Group (COG) studies of intermediate- and high-risk disease addressed the strength of early PET obtained after two cycles of chemotherapy; however the results are still pending.
Several cooperative group studies have focused on modifications of regimens to avoid gender-specific toxicities. The COG evaluated a strategy of stratifying patients with high-risk disease based both on response and gender. All patients received escalated bleomycin, etoposide, doxorubicin (Adriamycin), cyclophosphamide, vincristine, procarbazine, and prednisone (BEACOPP) therapy for 4 cycles. Patients with a rapid early response, defined as a 70% tumor reduction and having a negative gallium scan after four cycles, were then assigned to subsequent therapy based on gender. Boys received two additional cycles of ABVD designed to minimize risks of infertility and involved field radiation. Girls received four cycles of COPP/doxorubicin (Adriamycin), bleomycin, and vinblastine (ABV) and no radiation in order to avoid the risks of secondary breast cancer. Those with disease that met the criteria for slow early response received an additional 4 cycles of BEACOPP. Seventy-seven percent of girls avoided radiotherapy, whereas 68% of boys avoided alkylating agents during consolidation. The EFS and OS rates remained excellent.
The German GPOH-HD2002 study also evaluated sex-based modifications of therapy but did not use patient response for treatment allocation. Boys received two cycles of dose-intensified vincristine, etoposide, prednisone, and doxorubicin (Adriamycin; OEPA) therapy followed by two (for those with intermediate-risk disease) or four (for those with high-risk disease) cycles of cyclophosphamide, vincristine (Oncovin), prednisone, and dacarbazine (COPDAC) designed to attempt to limit gonadal toxicity. Girls received the same number of cycles but of OPPA followed by COPP. EFS and OS rates did not differ between the two sexes with the regimens employed.
The Pediatric Oncology Group (POG) evaluated response-based modifications of therapy without modifications for sex in patients with intermediate- and high-risk disease. In this study patients were evaluated for response after three cycles of therapy. Those with a rapid early response defined as a greater-than-70% reduction in disease and gallium negative, received 21 Gy IFRT. Those who were slow responders received an additional two cycles followed by radiation. With this strategy the 5-year EFS rate was the same in both groups, and the OS rate was 95%.
Whether radiotherapy can be omitted from treatment for at least some children and adolescents with Hodgkin lymphoma is one of the important questions in recent and ongoing clinical trials. Three cooperative group trials in the low-risk group each evaluated the removal of IFRT in a subset of patients with largely successful results. In the Stanford-St.Jude-Boston protocol, patients with a complete response, defined as greater than 75% reduction of the sum of the perpendicular of all lesions after two cycles of vinblastine, doxorubicin (Adriamycin), methotrexate (methotrexate), and prednisone (VAMP) chemotherapy were nonrandomly assigned to no radiation. All patients received four cycles of VAMP chemotherapy. Fifty three percent of patients met criteria for complete response. The 2-year EFS rates of this group were no different from those who did receive radiation, at 89% and 92% respectively. The GPOH-HD 2002 study also nonrandomly assigned patients at low risk with a complete response after two cycles, defined as more than 95% volume reduction, to no radiation. Thirty three percent of patients met this criterion, did not receive radiation, and had an outcome identical to those who did receive IFRT, with EFS rates at 5 years of 93% and 91% respectively. The Children’s Cancer Group (CCG) study 5942 randomly assigned patients who obtained a complete remission (CR), defined as 70% mass reduction of tumor volume and negative gallium scan, to radiation or no radiation after four cycles of COPP/ABV therapy. The EFS rates for randomized patients at 3 years were significantly different at 97% versus 91%; however, the OS rate at 3 years in both groups was 100%.
Omission of radiation has also been investigated in subgroups of patients with intermediate- and high-risk disease with mixed results. In the POG trial 8725, patients with stages IIB, IIIA2, IIIB, and IV were randomized to receive eight alternating cycles of MOPP/ABVD chemotherapy followed by total nodal irradiation (21 Gy) or no radiotherapy in cases of CR. There was no difference in EFS or OS between patient groups who did or did not receive radiation. In the GPOH-HD95 trial patients who achieved a complete response received no further therapy, whereas those who did not received 20 to 35 Gy of IFRT. In the intermediate- and high-risk groups, omission of radiation was associated with a significantly decreased EFS rate of 77% for those treated with chemotherapy only versus 92% for those who received combined modality therapy.
The CCG5942 trial included subjects with all disease stages. Based on their risk-group assignment, patients received either four or six cycles of COPP/ABV or six cycles of the mutiagent ABC regimen chemotherapy. Patients with at least a 70% tumor-volume reduction and negative gallium scan at the completion of chemotherapy were randomized to receive or not receive IFRT with 21 Gy. This randomized trial was stopped because of a significantly higher number of relapses in the no-IFRT arm. Most of the relapses were at sites of initial disease. Patients who did not receive IFRT had an EFS rate at 3 years from the time of randomization of 87% compared with 92% ( P = .057) for patients treated with IFRT in the intent-to-treat analysis. Importantly, longer term follow-up study of this cohort has shown that the OS in the two arms is no different. At 10 years postrandomization, the EFS of those treated or not treated with IFRT were 91.2% and 82.9% ( P = .004), respectively, whereas the OS of the two groups were 97.2% and 95.5% ( P = .5), respectively.
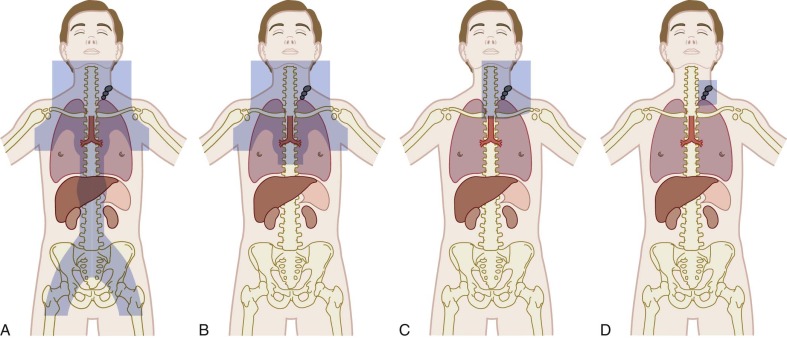
Radiation remains a critical component of therapy for the majority of children and adolescents with Hodgkin lymphoma. Radiotherapy planning involves decision making about both dosage and field, with the goals of maximizing efficacy but limiting late effects. Radiation fields have evolved over time ( Fig. 53-6 ). IFRT, used in the majority of contemporary protocols, generally involves radiation of the entire lymph-node region that includes the pathologically enlarged node. One major implication of the change from mantle-field radiation to IFRT for cervical disease is the exclusion of the axilla, which significantly reduces the exposure of lung and breast tissue. Investigations of further limitations to radiation fields are ongoing. The dosages in most contemporary studies used in combination with chemotherapy are generally no greater than 21 Gy, with some studies using higher dosages to sites of bulky or residual disease.
Disease Surveillance after Completion of Therapy
Driven both by required studies as part of clinical trials and as part of standard of care in some pediatric oncology settings, children and adolescents routinely undergo surveillance imaging with CT scans at regular intervals after completion of therapy. Given increasing awareness of the potential risks secondary to imaging-related radiation exposure, recent work has examined the utility of CT imaging as compared with clinical findings in the detection of disease recurrence. A retrospective review of patients treated in a POG study showed that the majority of relapses occurred within the first year and were detected by clinical findings. For the patients with late relapse, detection by either by clinical symptoms or surveillance imaging did not change the OS rate. These authors suggest limiting surveillance-CT imaging to the first 12 months after completion of therapy. FDG-PET scanning, which has utility in staging newly diagnosed patients and in the assessment of disease response (see above) has no established role in off-therapy surveillance. More widespread use of MRI as a replacement for CT may also allow for reduction of radiation exposure in lymphoma patients in the future.
Nodular Lymphocyte-Predominant Hodgkin Lymphoma
Pathologic Features
NLPHL is a monoclonal B-cell neoplasm. The histology is characterized by effacement of the lymph node architecture by a nodular or nodular and diffuse infiltrate of small lymphocytes, with an associated follicular dendritic network and scattered or clustered large cells referred to as lymphocytic and histiocytic (L&H) cells . NLPHL corresponds to Hodgkin paragranuloma of the classification of Jackson and Parker from 1947 and to lymphocyte-predominant Hodgkin lymphoma of the Rye modification of Lukes and Butler. Differentiation of NLPHL from the lymphocyte-rich subtype of classic Hodgkin lymphoma can be difficult in some cases. However, the phenotype of L&H cells (also termed popcorn cells because of their often multilobed nuclei) is different than classic Reed-Sternberg cells of the CHL subtypes in that they express CD45 and usually retain most normal markers of B-cell differentiation, such as CD20, CD79a, and the B-cell specific transcription factors BOB.1 and Oct-2, but do not express CD30 and CD15 (see Table 53-1 ). Also, markers of EBV infection are consistently absent from L&H cells of NLPLH. In some patients with NLPHL, progressively transformed germinal centers are observed in association with NPLHL. However, it remains uncertain whether these lesions are truly preneoplastic, because most patients with such lesions in reactive lymph-node hyperplasia do not develop a lymphoma.
Clinical Features
NLPHL comprises between 5% and 8% of Hodgkin lymphoma in children and adolescents and accounts for an even higher percentage of prepubertal patients. Unlike in patients with CHL, there is a significant male preponderance in those with NLPHL. The disease course is more indolent, and a higher proportion of patients have low-stage disease, most often presenting as localized peripheral adenopathy, most commonly in the neck and less commonly in the inguinal area. Mediastinal involvement is relatively rare. Unlike CHL, where contiguous nodal spread is the norm, in NLPHD noncontiguous spread is observed in those with high-stage disease. The pattern of relapse of disease also differs from CHL, with relapses occurring late and often repeatedly. An additional unique feature of NLPHL is the increased risk of transformation to NHL occurring in 5% to 12% of cases, whereas this is very rarely observed in CHD.
Diagnostic evaluation should include excisional node biopsy. Fine-needle aspirate is often inadequate and can lead to the misdiagnosis of reactive nodal tissue. Staging of patients with NLPHL uses the Ann Arbor system as used for CHL, and staging evaluations to be considered are the same as those for a patient with CHL except that bone marrow biopsies are rarely indicated. The only consistent prognostic factor for patients with NLPHL is stage of disease, although some data suggest that a ratio of lymphocyte count to monocyte count at the time of diagnosis may also have prognostic value.
Treatment and Outcomes
Historically patients with NLPHL have been included in most therapeutic studies of childhood and adolescent Hodgkin lymphoma. Uncertainty remains about optimal therapy for these patients. Given the favorable prognosis and the burden of late effects from therapy, the evaluation of less intensive regimens has been undertaken.
The majority of patients with NLPHL have low-stage disease, which is sometimes amenable to complete resection. One of the unique features of NLPHD is that a portion of children with low-stage disease treated with resection alone are cured of their disease. Studies of patients treated in this fashion have reported relapse-free survival rates of 67% to 100%. All of the recurrences in patients with relapse were low stage, and the OS has been reported as 100%. For pediatric patients with NLPHD, a watchful waiting approach for those who are in CR after initial surgery is appropriate. However, a careful postoperative staging and meticulous follow-up observation is essential. Other strategies for patients with low-stage disease are the use of radiotherapy alone or low-intensity alkylator-based chemotherapy regimens.
Data to evaluate the best therapy for patients with advanced stage NLPHL are relatively scarce. Outcome of these patients is similar to those with CHL, and the usual recommendation is that they receive therapy similar to those with advanced-stage CHL. Investigation of the possible role for therapy with rituximab as a single agent or as part of combination therapy for this disease are ongoing.
Treatment of Relapsed Hodgkin Lymphoma
Although the success of primary therapy for children and adolescents is high, approximately 10% of patients develop relapsed disease. The outcome for those with relapsed disease is variable. The two strongest predictive factors are time to relapse and response to reinduction chemotherapy. Time to relapse can be subdivided into three prognostic groups: 1) patients with primary refractory disease or recurrent disease within the first 3 months have the worst outcomes; 2) those with disease relapse within 12 months of diagnosis form an intermediate group; and 3) those with relapses occurring longer than 12 months from diagnosis have the most favorable outcome. Patients who have no response to reinduction therapy have a dismal prognosis, as do those with active disease immediately before autologous stem cell transplantation (SCT). Additional prognostic factors include the intensity of primary therapy, stage, and B symptoms at relapse.
There is no consensus on optimal salvage therapy for children with relapsed disease. The options include chemotherapy with or without radiation, autologous SCT, allogeneic SCT, or novel therapies. Data to support treatment choices can be difficult to interpret because there is a lack of randomized trials evaluating these therapeutic options in children; the majority of studies in patients with relapse have been single-arm trials with varying inclusion criteria.
Patients with late relapse who have responsive disease to reinduction therapy have a favorable prognosis with treatment with chemotherapy and radiation. Chemotherapy regimens have been evaluated in this setting including ifosfamide, carboplatin, and etoposide (ICE) ; carmustine (BCNU), etoposide, cytarabine (ara-C), and melphalan (miniBEAM) ; cytarabine, cisplatinum, and etoposide (APE) ; as well as newer regimens including gemcitabine and vinorelbine (GV) among others. Information that should be considered in choosing a salvage regimen should include knowledge of the frontline therapy used, in part to avoid toxicities with cumulative drug exposure.
For patients with high-risk relapsed disease, intensifying therapy with autologous SCT should be considered, although controversy about which patients are likely to benefit from this therapy remains. In a prospective randomized trial of the German Hodgkin Lymphoma Study Group, adults with chemotherapy-sensitive disease had a significantly higher failure-free survival probability when treated with two cycles of dexamethasone, carmustine (BCNU), etoposide, cytarabine (ara-C), and melphalan (Dexa-BEAM) followed by autologous SCT compared with patients receiving four cycles of Dexa-BEAM without SCT. For childhood Hodgkin lymphoma there are very few prospective or randomized trials. In a retrospective study of 51 pediatric patients with recurrent Hodgkin lymphoma treated with different salvage regimens, there was a beneficial impact of autologous SCT on survival only for patients refractory to first-line therapy, but sensitive to second-line therapy disease. The outcome of children with disease that is unresponsive to chemotherapy who undergo autologous SCT is dismal.
The European trial for children with relapsed Hodgkin lymphoma, EuroNet-PHL-C1, divides patients into low-, intermediate- and high-risk groups based on time to relapse and early response assessment. Patients with low-risk disease are treated with chemotherapy and radiation. Those with high-risk disease have their therapy intensified with autologous SCT. Those with intermediate-risk disease have therapy allocated based on response, with those with a good response receiving chemotherapy and radiotherapy only.
The use of allogeneic transplant in relapsed Hodgkin lymphoma, with the hopes of making use of graft-versus-lymphoma effects has been investigated including myeloablative SCT, reduced intensity SCT, and reduced intensity SCT after autologous SCT. In this very–high-risk group, both relapsed disease and transplant-related mortality limit the adoption of this therapy except for those patients with adequate performance status and limited curative options.
Novel therapies for relapsed disease are needed. One agent that shows significant promise is the antibody-drug conjugate brentuximab vedotin (also known as SGN-35 ), which combines an anti-CD30 monoclonal antibody with the antimicrotubule agent, monomethyl auristatin E. In a phase 2 study of patients with very–high-risk disease (71% had primary refractory disease, and 100% of patients had previously been treated with autologous SCT), the overall response rate was 75%, with 34% of patients achieving a CR and with a high percentage of these patients remaining without evidence of relapsed disease at 1.5 years. The median age of the patients in this trial was 31 years, with the youngest being 15 years old. Toxicity of the agent was relatively mild, including neuropathy and cytopenias. The safety and effectiveness of adding this agent to standard chemotherapy as well as including it in front-line therapy is being investigated by several cooperative groups.
Late Effects of Therapy
Given the high rate of cure in children and adolescents with Hodgkin lymphoma, the risks of long-term effects of treatment are very important. In the scientific literature, the number of reports on late sequelae after successful treatment of pediatric Hodgkin lymphoma patients has exceeded that of reports on prospective therapeutic trials. Many of these reports document late sequelae associated with earlier treatment approaches. The treatment for pediatric Hodgkin lymphoma has evolved because of a continuous process of attempting to maximize efficacy while limiting the long-term risks of therapy. Therefore many of the reports of sequelae of therapy administered several decades ago may not accurately reflect the risks of late effects associated with current treatment strategies.
Some late effects of Hodgkin lymphoma therapy can be attributed to a distinct treatment modality or a single drug; others may have a multifactorial pathogenesis. Table 53-10 lists late effects that have impact on morbidity and mortality, as well as those that affect the quality of life for survivors.
| Late Effect | Radiotherapy | Chemotherapy |
|---|---|---|
| Musculoskeletal growth impairment | X | |
| Cardiomyopathy | Anthracyclines | |
| Coronary artery disease, valvular disease, and pericardial disorders | X | |
| Stroke | X | |
| Infections (postsplenectomy or splenic radiation) | X | |
| Thyroid dysfunction | X | |
| Female gonadal dysfunction | X | |
| Male infertility | Inguinal, pelvic irradiation | Alkylating agents; procarbazine |
| Lung dysfunction | X | Bleomycin; BCNU; CCNU; busulfan |
| Second cancer (hematologic) | Alkylating agents; topoisomerase II inhibitors | |
| Solid tumors | X |
In two large, single-center series of patients with pediatric Hodgkin lymphoma treated over several decades, the long-term cumulative risk of death from treatment-related sequelae such as infections, cardiac disease, and second cancers approached that of the risk of death from relapsed Hodgkin lymphoma. Data from 2742 patients treated between 1970 and 1986 who were under the age of 21 at the time of diagnosis, who were alive longer than 5 years posttherapy, and who were in the Childhood Cancer Survivor Study (CCSS) has delineated the risks and causes of excess mortality. With a median follow-up length of greater than 20 years, of the 500 observed deaths in the cohort, 35% were from Hodgkin lymphoma, 23% from second malignant neoplasms (SMNs), and 14% from cardiovascular disease. The 30-year OS rate from diagnosis was 74%.
SMNs are devastating sequelae of therapy for Hodgkin lymphoma. In the U.S. Childhood Cancer Survivor Cohort consisting of more than 13,000 survivors of childhood cancers, patients treated for Hodgkin lymphoma were at the highest risk to develop an SMN. Patients treated with full-dose radiation are at significant risk for SMNs with a cumulative incidence of greater than 25% after 30 years of follow-up study. Fifteen years postdiagnosis of Hodgkin lymphoma, the second leading cause of mortality after death from the primary disease is death from an SMN. Breast cancer makes up the largest component of the excess solid tumors in female patients.
Contemporary therapy no longer uses high-dose extended-field radiation. The impact of lowering radiation dosage and the use of combination chemotherapy on risks of SMN continues to be investigated. In a cohort study including both children and adults treated for Hodgkin lymphoma, 459 second malignancies were described in 5798 patients. These patients were treated between 1963 and 2001. All received chemotherapy, and 3432 also received radiation. Of those who received chemotherapy only, there remained significantly increased risks of second malignancies including leukemia, NHL, and lung cancer with the peak period of risk between 5 and 9 years posttherapy. For those who received combined modality therapy including radiation, the absolute risk of an SMN was significantly higher and included a wider arrays of malignancies; the period of risk extended to 25 years and longer. In a small cohort of 112 pediatric patients treated at a single institution between 1970 and 1990 with combined modality therapy including 15 to 25.5 Gy IFRT, the rate of SMN including sarcomas and breast and thyroid carcinomas was similar to that reported in studies of patients who had received higher doses of radiation. Other studies have suggested that low-dose radiation and limiting cumulative alkylator exposure may have a protective effect in adult patients but not in children. Still others have shown that patients who received less than 23 Gy of mediastinal radiation had a lower incidence of breast cancer, which may in part be related to the transition from extended-field radiation to IFRT. The cumulative incidence of second leukemias ranges from 0.6% to 2.1% and reaches a plateau after 10 to 14 years. The risk of second leukemias is associated with the cumulative dose of alkylating agents and is almost absent in patients treated with radiotherapy alone. The risk of developing thyroid cancer is clearly related to radiotherapy and is highest in young children. Almost all thyroid tumors arise within the radiation field. A recent study analyzing the genetic factors that may contribute to radiation-induced tumors in survivors of Hodgkin lymphoma has identified variants at chromosome band 6q21, implicating the PRDM1 gene in the etiology of secondary malignancies.
Cardiovascular disease contributes significantly to the morbidity and mortality of patients who are survivors of Hodgkin lymphoma. Damage to the heart after full dose radiotherapy includes coronary artery disease, valvular pathology, and pericardial disease. Cardiomyopathy with ventricular dysfunction can result from anthracycline therapy. Risks of late anthracycline cardiotoxicity are higher in young children, in girls, and with higher cumulative doses. In long-term follow-up observation of a cohort of patients from a single-center combination of exposure to mediastinal radiation and anthracycline was associated with the highest risk of cardiac morbidity. Data regarding the long-term cardiac risks associated with contemporary therapy using low-dose IFRT and chemotherapy regimens that generally limit cumulative anthracyclines exposure to less than 250 mg/m 2 are needed.
Death from overwhelming infections related to splenectomy was a major risk that has been significantly reduced since splenectomy was abandoned from staging procedures. Irradiation of the spleen may also induce functional impairment of the spleen, at least if dosages of 20 Gy or more are used.
Chronic lung disease can result from irradiation of the lung and exposure to several chemotherapy agents used in Hodgkin lymphoma regimens, including bleomycin and the nitrosourea derivatives BCNU and lomustine (CCNU). The cumulative dose of these drugs is the most crucial determinant for chronic lung injury. Very young age may be an additional risk factor for possible fatal lung toxicity from BCNU and CCNU. The combination of radiotherapy and drugs with lung toxicity contributes to increased risk of long-term lung impairments. This is of special concern in patients requiring salvage treatment for recurrent Hodgkin lymphoma that includes high-dose chemotherapy.
Survivors of pediatric Hodgkin lymphoma are at increased risk for thyroid malfunction—mainly hypothyroidism but also hyperthyroidism, benign and malignant thyroid nodules, and Graves disease. The risk increases with the dose of neck irradiation, younger age at treatment, and female gender.
Musculoskeletal growth impairment was one of the first sequelae recognized to be a consequence of high-dose radiotherapy for children with Hodgkin lymphoma. Prepubertal children treated with 40 Gy of mantle irradiation would have predictable sequelae of lack of growth of the bone and soft tissues of the neck and clavicles, leading to orthopedic complications as well as significant cosmetic effects. This complication is ameliorated by low-dose IFRT used in most contemporary regimens.
The primary risk factor for gonadal toxicity is exposure to alkylating agents, primarily cyclophosphamide and procarbazine. Historically mechlorethamine was the primary cause of chemotherapy-related sterility in males. Dose-dependent testicular damage from germ-cell depletion and Leydig-cell dysfunction has also been documented in prepubertal and postpubertal boys after MOPP; chlorambucil, vinblastine, procarbazine and prednisone (ChlVPP); and OPPA-COPP chemotherapy. The ABVD regimen appears to be less toxic to germ cells. Results from the German-Austrian studies on pediatric Hodgkin lymphoma have shown clearly that with moderate doses of cyclophosphamide, male infertility correlates with the cumulative dose of procarbazine. Historically in girls, the most substantial risk of infertility was from pelvic lymph node irradiation and was ameliorated to some extent by ovarian transposition. The risks of early menopause for females receiving contemporary therapy is likely fairly low, and small studies have shown no measurable impact of ABVD therapy on fertility in women.
Follow-up care of survivors of Hodgkin lymphoma should follow a structured plan such as that described in the COG Long-Term Follow-up Guidelines. Availability of a detailed and accurate treatment summary is imperative to determining appropriate care and surveillance investigations in follow-up observation.
Non-Hodgkin Lymphoma
NHLs are a diverse collection of malignant neoplasms of lymphoid cell origin, including all the malignant lymphomas that are not classified as Hodgkin lymphoma. They account for 7% of all cancers in those younger than 20 years old. NHLs are heterogeneous in their pathology, clinical features, and responsiveness to therapy. With improved understanding of biology and data from sequential disease-based clinical trials as well as better supportive care, the outcome of children with NHL has improved dramatically over the last several decades. With conventional therapy, depending on subtype and stage, the majority of children with NHL can be cured of their disease.
The classification of NHL has been and continues to be a topic of scientific discussion and a source of confusion for clinicians. The Rappaport system used growth patterns (diffuse vs. nodular) and cytology (undifferentiated vs. differentiated) as the basic criteria for disease definitions. The Lukes Collins classification system was based primarily on splitting the lymphocytic system into T and B lymphocytes. The Kiel classification system attempted to translate information about how the lymphatic system is organized into a classification system for NHL, in which the lymphoma cells were related morphologically and immunophenotypically to the normal cell categories of the immune system. The National Cancer Institute Working Formulation for Clinical Usage, established in 1982, did not intend to constitute another classification of NHL; rather it was to be a common language to translate between existing classifications. It took into consideration the clinical course of patients and resulted in a grading of NHL into low-grade, intermediate-grade, and high-grade NHL. This system was widely used by clinicians in North America. In Europe and Asia, the Kiel classification was more broadly used. The use of different classification systems hampered or even made it impossible to compare results of clinical trials. In the 1990s, the International Lymphoma Study Group undertook a major effort to overcome this confusion and to create a uniform classification of NHL based on biologic principles. The result was the Revised European-American Classification of Lymphoid Neoplasms (REAL) classification, which then lead to the WHO Classification of Tumors of the Hematopoietic and Lymphoid Tissues produced in 2002 and revised in 2008. The basic principles of the WHO classification system include subdivision of neoplasms according to the lineage (B, T, or natural killer [NK] cell) and the definition of distinct subtypes within each lineage according to a combination of morphology, immunophenotype, genetic features, and clinical features.
The revisions of the WHO classification system in 2008 included diseases that overlap between entities, such as B-cell lymphomas with features intermediate between DLBCL and BL and B-cell lymphomas with features intermediate between DLBCL and Hodgkin lymphoma. In addition, age-specific factors defined several new disorders including pediatric type follicular lymphoma (FL) and systemic EBV-positive T-cell lymphoproliferative disease of childhood.
Combination chemotherapy is the cornerstone of successful treatment of children and adolescents with NHL. A key moment in the development of current treatment concepts was the recognition that different NHL subtypes require different chemotherapeutic strategies. In children and adolescents with NHL, three main disease groups account for the vast majority of the diagnoses. The three are LL, mature B-cell neoplasms (including BL and DLBCL), and anaplastic large-cell lymphoma (ALCL; Fig. 53-7 ). In the era of solid organ and SCT, posttransplant lymphoproliferative diseases are becoming more common.

Epidemiology and Causative Factors
There is significant variation in incidence rates according to geographic areas, gender, age, and ethnicity in childhood NHL. The comparison of incidence rates of NHL in childhood and adolescence in different geographic areas is limited because of a paucity of true population-based registries, different degrees of completeness of data, and the use of various disease classification systems over time. The reported incidence rates range from 6.6 per million population in the United Kingdom to 12.5 per million in Japan. Specific subtypes of NHL, however, occur at a much higher rate in particular regions. Endemic BL accounts for 74% of all childhood malignancies in equatorial Africa, whereas all NHLs combined account for 7% of pediatric cancer in North America.
In all registries the incidence rates of NHL are higher in males than in females, usually by a factor of 2. The male-to-female ratio varies considerably among different NHL subtypes. Whether male predominance in most NHL subtypes reflects hormonal differences rather than loss of an X-chromosome–linked tumor suppressor gene remains to be determined. The incidence of NHL and of the various subtypes varies widely by age, with substantial differences between children and adults. Among patients younger than 18 years of age there is also considerable variability ( Fig. 53-8 ). NHL as a whole in children younger than 3 years of age is extremely rare. BL or leukemia is the predominant subtype in younger children. The incidence of DLBCL and ALCL increases in those older than 15 years of age.
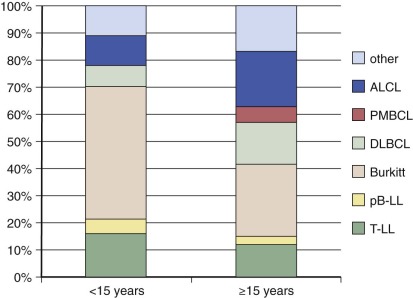
In the U.S. Surveillance, Epidemiology, and End Results (SEER) program, the incidence rate in Caucasian children is higher as compared with African American children. In adults, an increased risk for NHL has been described in first-degree relatives of patients with hematopoietic malignancies. Except for classic known hereditary immunodeficiency syndromes predisposing for NHL, only a few specific genes have been associated with risks of childhood NHL. Biallelic mutations in mismatch repair genes have been associated in a series of families with several types of childhood malignancies including T-cell lymphomas.
Conclusive data to support the association of environmental factors and risk of childhood NHL are extremely limited. Conversely, data to support the role of infection in a subset of NHLs has been long established. Denis Burkitt not only recognized and described BL as a new disease entity but also established the epidemiologic basics for the current view of its association with EBV, malaria, and possibly arbovirus infections and described the lymphoma belt of Africa.” In 1964, Epstein and associates described viral particles—later referred to as EBV—in BL samples from African patients. Subsequently the EBV genome was found to be incorporated into 96% of endemic BL cases in the African lymphoma belt, whereas in France and Germany, only 10% to 15% of cases are EBV-positive. It was postulated that EBV infection in addition to exposure to malaria and possibly arboviral infections early in life may be crucial for the pathogenesis of endemic EBV-positive BL. In addition to endemic BL, lymphoproliferative disease, and lymphomas in children with acquired or congenital immunodeficiency are often EBV positive.
Inherited and acquired immunodeficiency is a strong risk factor for developing NHL during childhood. Table 53-11 presents the most common inherited immunodeficiencies associated with lymphoproliferative disorders and lymphoma. For patients with these inherited (also called primary ) immunodeficiencies, the chance of dying of lymphoma is 10 to 200 times the expected rate for age-matched unaffected children. Many of the lymphomas in this patient group are EBV-positive. There is a male preponderance, partly explained by the fact that some of the inherited immunodeficiencies are X-linked disorders. Children infected with human immunodeficiency virus (HIV) have more than 150 times the risk of developing NHL in comparison with general population rates. NHLs are the most common neoplasms occurring in children with HIV, and most are of B-cell lineage. For some children NHL may be the first defining condition for acquired immunodeficiency syndrome (AIDS). The introduction of highly active antiretroviral therapy has been associated with a significant decrease the incidence of some HIV-related malignancies, such as Kaposi sarcoma, but the effect on the incidence of NHL appears to be more modest.
| Syndrome | Genetics/Pathogenesis | Immune Defect | Malignancy |
|---|---|---|---|
| Ataxia telangiectasia | ATM ; impaired DNA repair | Progressive decline in T cells, abnormal immunoglobulins | T-cell ALL and T-cell LL, CHL, BL, DLBCL |
| Wiskott-Aldrich syndrome (WAS) | WASP ; abnormal cytoskeletal architecture of hematopoietic cells | Defects in T cells, B cells, neutrophils, macrophages | EBV-associated DLBCL, Hodgkin, lymphomatoid granulomatosis |
| Severe combined immunodeficiency (SCID) | X-linked: gamma chain mutations with defective IL signaling; ADA : defective adenosine deaminase | Dependent of subtype but in general low or absent T and NK cells, nonfunctional B cells | EBV-associated lesions |
| X-linked lymphoproliferative disease (XLP) | SH2D1A ; SAP, which modulates B- and T-cell interactions | Defective EBV-specific T and NK cells | EBV-associated hematophagocytic lymphohistiocytosis, B-cell lymphoproliferative disease |
| Autoimmune lymphoproliferative syndrome (ALPS) | Defects in FAS -mediated apoptosis pathway | Increased CD4 and CD8 T cells | LPHL, CHL, DLBCL, BL |
| Common variable immunodeficiency (CVID) | Defects in genes encoding ICOS, CD19, BAFFR | Low IgG and IgA, decreased B cells | EBV-associated lesions, DLBCL, Hodgkin |
| X-linked hyperimmunoglobulin M syndrome | CD40 ligand mutation leading to defective B cells | Low or absent IgG and IgA, variable T-cell defects | EBV-associated lesions, DLBCL, Hodgkin |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


