Chapter Outline
HISTOPATHOLOGY OF GERM CELL TUMORS
EPIDEMIOLOGY OF PEDIATRIC GERM CELL TUMORS
MOLECULAR BIOLOGY OF GERM CELL TUMORS
Chromosomal Constitution and Copy Number Aberrations
Genome-Wide Association Studies
The Role of Developmental Signaling Pathways in Germ Cell Tumor Pathogenesis
DIAGNOSIS AND STAGING OF PEDIATRIC GERM CELL TUMORS
Surgical Approach to Sacrococcygeal Tumors
Surgical Approach to Testes Tumors
Surgical Approach to Ovarian Tumors
Surgical Approach to Mediastinal Germ Cell Tumors
PROGNOSTIC FACTORS AT DIAGNOSIS OF PEDIATRIC GERM CELL TUMORS
Explanation of the Nomenclature
Infantile Choriocarcinoma: A Rare but Rapidly Fatal Condition if Not Recognized
TREATMENT OF STAGE I MALIGNANT NONSEMINOMATOUS GERM CELL TUMORS
Chemotherapy for Pediatric Germ Cell Tumors (> Stage I)
Treatment of Pediatric Germ Cell Tumors in the United States
Treatment of Pediatric Germ Cell Tumors in the United Kingdom
Treatment of Pediatric Germ Cell Tumors in Germany
The Dose of Bleomycin in Treatment of Pediatric Germ Cell Tumors
Late Effects Among Men Treated for Testicular Cancer
Cisplatin Levels are Measurable for Years After Treatment
Risk of Cardiovascular Disease
Risk of Second Malignant Neoplasm
Germ cell tumor (GCT) is the designation given to neoplasms arising from the cells of the germline—the cells that are destined to become either the egg or the sperm. A number of unique features of these tumors including their bimodal and wide age distribution, remarkable phenotypic diversity, and varying biologic behavior make GCTs a particular challenge for the surgeon, pathologist, and oncologist. Successful treatment regimens developed over the last several decades have focused current clinical research on ways to maintain efficacy and minimize toxicity for most patients and intensify treatment for patients who fail first-line therapy. Recent advances in understanding the underlying aberrations in germline development shed light on the genesis of these tumors and may provide insight into new avenues for treatment.
Development of the Germline
The pathogenesis of GCTs can best be understood through an analysis of the mechanisms of germline development. The role of the germ cell is to ensure the continuation of a species by producing the gametes, cells that will give rise to the next generation. Reflecting this unique role, germ cells are set aside from somatic cells very early in development. In humans, germ cells arise in an extraembryonic position and must migrate to the site at which the gonads will form. The pluripotency of germ cells is reflected in the wide histopathologic diversity of GCTs.
Specification of Primordial Germ Cells
Much of what is known about early molecular events in the mammalian germline comes from examination of germ-cell development in the mouse. Similar molecular mechanisms of germ cell development appear to operate in humans, based on studies of human embryos and human embryonic stem cells differentiating into germ cells in vitro. Development of the germline begins at the time of blastocyst implantation, when the extraembryonic ectoderm and the visceral endoderm send signals to cells in the proximal epiblast, also known as the embryonic ectoderm ( Fig. 63-1 ). The major inductive signals are the bone morphogenetic proteins (BMPs), which are members of the transforming growth factor β (TGF-β) superfamily. In response to BMPs, some of the epiblast cells begin to express the marker fragilis , signaling their competence to become germ cells. Of these fragilis -expressing cells, a few begin to express the transcriptional repressor Blimp1/Prdm1 . These cells, in which expression of somatic genes such as Hoxb1, T/Brachyury, and Snail is repressed, will become the primordial germ cells (PGCs). Unlike other epiblast-derived cells, PGCs regain or maintain expression of certain genes associated with pluripotency such as STELLA, OCT3/4, and NANOG . Certain pluripotency genes can be reactivated in GCTs and may contribute to malignant potential; for example, NANOG and OCT3/4 have been used as sensitive markers of malignant germ cells in studies of GCTs. In humans, PGCs can be identified in the wall of the yolk sac by their intrinsic alkaline phosphatase activity beginning at about day 24. The PGCs begin to proliferate as they migrate out of the yolk sac into the embryo.
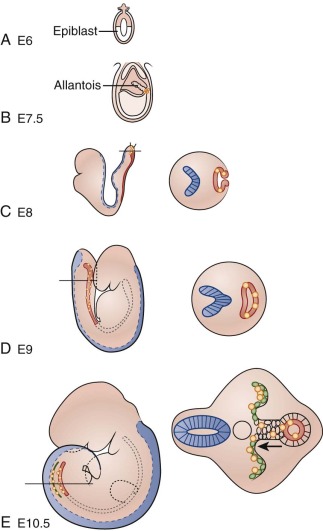
Primordial Germ Cell Migration
In humans, as in many other organisms, the PGCs arise in an extraembryonic location, distant from the eventual site at which the gonad will form. This physical separation may serve to insulate the germ cells from various proliferation and differentiation signals in the developing embryo or to enforce a quality control by selecting for healthy PGCs capable of successfully navigating to the developing gonad. Proper PGC migration is critical to the survival of the germ cells and formation of the gonad, and failure of this migration can result in ectopic germ cells. Persistence and malignant transformation of these ectopic germ cells is one possible mechanism by which extragonadal GCTs are thought to arise.
Toward the end of gastrulation, morphogenetic movements in the developing embryo bring the PGCs in proximity to the hindgut. Invading the endoderm, the PGCs colonize the hindgut and begin vigorous, apparently random migratory movements, remaining confined to the gut. The receptor tyrosine kinase c-kit, expressed in PGCs, and its ligand, Steel, expressed in somatic cells, are required for the colonization of the hindgut and for the survival and migration of PGCs within the gut. Wylie and co-workers have shown that in mice, PGCs deficient in c-kit signaling undergo apoptosis through the action of the p53-target gene, Bax . At 5 to 6 weeks after fertilization, the PGCs upregulate expression of the adhesion factor E-cadherin and exit the hindgut on the dorsal side to begin colonization of the genital ridge primordia. The genital ridges are bilateral swellings of mesenchyme covered by coelomic epithelium and situated on either side of the dorsal aorta on the posterior wall of the embryo in the lower thoracic and lumbar regions. The genital ridges appear to attract the PGCs because of their expression of stromal cell–derived factor 1 (SDF-1, or CXCL12), which is a ligand for the chemokine receptor CXCR4, which is expressed in the PGCs. As the PGCs exit the hindgut, they divide into two lateral streams to colonize the genital ridges. PGCs that fail to colonize the ridges and remain in the midline are eliminated by apoptosis, as the midline cells downregulate expression of Steel. Once they have entered the genital ridges, PGCs become much less motile but continue several more rounds of division. This proliferation is dependent on continued Steel/c-kit signaling.
Erasure of Imprinting
Imprinting refers to the epigenetic modification of certain genes (typically by cytosine methylation) such that only the maternal or paternal allele of the gene is expressed. Lineage-specific patterns of imprinting are established in different tissues, including the germline, around the time of gastrulation. Upon entering the gonadal ridges, however, PGCs actively erase these genomic methylation patterns. This erasure is necessary in order to allow the maternal and paternal imprinting patterns to be established in the oocytes and sperm, respectively. Imprinting is reestablished in sex-specific patterns during gonadogenesis. GCTs exhibit partial or total erasure of imprinting, implying their origin from early germ cells.
Gonadogenesis
Beginning shortly before the arrival of the PGCs in the genital ridges, the coelomic epithelium begins to proliferate and invade the underlying mesenchyme, forming the primitive sex cords. Migrating PGCs entering the gonad are surrounded by the cords. At this early stage, the appearance of the developing gonad is identical in males and females, and the tissue is referred to as the indifferent gonad. Subsequently, changes occur in both the germ cells and the gonadal somatic cells according to the genetic sex of the embryo. In genetic males, sex determination occurs under the influence of the testis-determining SRY gene on the Y chromosome. After the initial rounds of cell division, male germ cells enter a mitotic arrest that persists until after birth. In males, the primitive sex cords proliferate and penetrate deeper into the mesenchyme, forming the testis or medullary cords, which connect proximally to form the rete testis. By the fourth month of development, the cords consist of germ cells and the supporting Sertoli cells, which are derived from the surface epithelium of the genital ridge. The cords remain solid until puberty, when they form lumens to become seminiferous tubules. By the eighth week of development the mesenchyme of the gonadal ridge gives rise in males to Leydig cells, which secrete testosterone. Secretion of antimüllerian hormone by the Sertoli cells and testosterone by the Leydig cells leads in males to degeneration of the paramesonephric ducts and the development of the mesonephric ducts into the vas deferens and epididymis. Testosterone is also necessary for male differentiation of the external genitalia.
Female gonadal development proceeds along different lines. In the absence of the Y chromosome, the sex cords initially degenerate and are replaced by a new set of cortical sex cords derived from the surface epithelium. By the fourth month the cortical sex cords have become isolated clusters with each cluster surrounding a single germ cell. In females the primitive germ cells, called oogonia, continue rapid proliferation, reaching maximal numbers by the seventh month. After that point, most of the oogonia degenerate; the remaining cells, now called primary oocytes, enter meiosis and arrest at the diplotene stage of the first meiotic prophase. Granulosa cells (derived from the surface epithelium) and thecal cells (derived from the mesenchyme) together form the follicle cells that surround each primary oocyte. Beginning at adolescence, groups of oocytes periodically resume meiosis. In females, the paramesonephric (müllerian) ducts develop into the oviducts, uterus, cervix, and upper part of the vagina. The mesonephric ducts degenerate.
In summary, development of the germline requires the proper specification of PGCs and their migration through the embryo to the gonadal ridges, where the gonads are formed through interactions of the germ cells with somatic cells. Concomitant with this process, the inherited pattern of genomic imprinting is erased, and a new, sex-specific pattern is formed. The complex process of gonadal organogenesis is subject to both genetic and environmental influences. Abnormal development of the gonads during the embryonic and fetal periods leads to defects such as cryptorchidism and gonadal dysgenesis, which are strongly associated with the risk of developing GCTs.
Histopathology of Germ Cell Tumors
GCTs are a heterogeneous group of neoplasms with a wide variety of histopathologic features. This variety reflects the pluripotent nature of the PGCs from which GCTs arise. Adding to the complexity of this tumor type, GCTs with apparently similar histopathology can have very different biologic behaviors when presenting at different ages or anatomic sites. The five major histologic subtypes of GCTs are teratoma, yolk sac tumor (YST), germinoma, embryonal carcinoma, and choriocarcinoma. The World Health Organization (WHO) classification of GCTs is presented in Boxes 63-1 and 63-2 .
Germ Cell Tumors
Intratubular germ cell neoplasia, unclassified
Other types
Tumors of One Histologic Type
Seminoma
Seminoma with syncytiotrophoblastic cells
Spermatocytic seminoma
Spermatocytic seminoma with sarcoma
Embryonal carcinoma
Yolk sac tumor
Trophoblastic tumors
Choriocarcinoma
Trophoblastic neoplasms other than choriocarcinoma
Monophasic choriocarcinoma
Placental site trophoblastic tumor
Teratoma
Dermoid cyst
Monodermal teratoma
Teratoma with somatic type malignancies
Tumors of More Than One Histologic Type
Mixed embryonal carcinoma and teratoma
Mixed teratoma and seminoma
Choriocarcinoma and teratoma/embryonal carcinoma
Others
Sex Cord/Gonadal Stromal Tumors
Pure Forms
Leydig cell tumor
Malignant Leydig cell tumor
Sertoli cell tumor
Sertoli cell tumor lipid rich variant
Sclerosing Sertoli cell tumor
Large cell calcifying Sertoli cell tumor
Malignant Sertoli cell tumor
Granulosa cell tumor
Adult type granulosa cell tumor
Juvenile type granulosa cell tumor
Tumors of the thecoma/fibroma group
Thecoma
Fibroma
Sex cord/gonadal stromal tumor, incompletely differentiated
Sex cord/gonadal stromal tumor, mixed forms
Malignant sex cord/gonadal stromal tumor
Tumors containing both germ cell and sex cord/gonadal stromal elements
Gonadoblastoma
Germ cell-sex cord/gonadal stromal tumor, unclassified
Germ Cell Tumors
Dysgerminoma
Yolk Sac Tumor
Polyvesicular vitelline tumor
Glandular variant
Hepatoid variant
Embryonal carcinoma
Polyembryoma
Nongestational choriocarcinoma
Mixed germ cell tumor
Biphasic Or Triphasic Teratoma
Immature teratoma
Mature teratoma
Solid
Cystic
Dermoid cyst
Fetiform teratoma
Monodermal Teratoma and Somatic-Type Tumors Associated with Dermoid Cysts
Thyroid (Struma ovarii)
Carcinoid
Neuroectodermal
Carcinoma
Melanocytic
Sarcoma
Sebaceous tumors
Germ Cell Sex Cord-Stromal Tumors
Gonadoblastoma
Variant with malignant germ cell tumor
Mixed germ cell sex cord-stromal tumor
Variant with malignant germ cell tumor
Sex Cord-Stromal Tumors
Granulosa-stromal cell tumor
Granulosa cell tumor
Adult type granulosa cell tumor
Juvenile type granulosa cell tumor
Tumors of the thecoma/fibroma group
Thecoma
Fibroma
Sertoli-stromal cell tumors
Sertoli cell tumor
Stromal-Leydig cell tumor
Sex cord-stromal tumors of mixed or unclassified types
Steroid-cell tumors
Leydig cell tumor
Steroid cell tumor, not otherwise specified
Overview of Testicular Germ Cell Tumors
Before puberty in infants and children, teratoma and/or YST account for the vast majority of GCTs. Seminoma (the term used for a testicular germinoma), embryonal carcinoma, and choriocarcinoma are very rare in this age group. GCTs of the prepubertal testis differ from their adult counterparts not only in the distribution of histologic subtypes, but also in their spectrum of cytogenetic abnormalities. After the early peak of testicular YST and teratoma in the 0-to-4–years age group, testicular tumors are relatively rare until the onset of puberty. Between ages 10 and 40 years, there is a second peak in testicular tumor incidence, comprised of seminoma and nonseminomas (including embryonal carcinoma, teratoma, YST, and choriocarcinoma). Postpubertal tumors demonstrate a wider array of histopathologic differentiation than do the GCTs of the prepubertal testis. Moreover, adolescent and adult GCTs share a common precursor cell of origin (the carcinoma in situ [CIS] or intratubular germ cell neoplasia, unclassified [IGCNU]), as well as characteristic cytogenetic abnormalities including the near-universal presence of amplifications of chromosome 12p. These histopathologic and molecular cytogenetic differences have lent support to the hypothesis that different pathogenic mechanisms underlie juvenile and adult testicular GCTs.
Overview of Ovarian Germ Cell Tumors
In neonates and infants, the most common ovarian lesions are ovarian cysts. Cysts can occur any time from late gestational stages onwards, and they occur at a greater rate in babies of mothers with diabetes, preeclampsia, and Rh immunization. Ovarian cysts in children often resolve spontaneously without intervention. GCTs account for 75% of ovarian tumors in the first 2 decades of life and up to 90% of ovarian tumors in premenarchal girls. The majority of these (95%) are benign, mature teratomas, mostly commonly “desmoid” tumors. Five percent of ovarian GCTs are malignant; these include dysgerminomas, immature teratomas, mature teratomas with somatic malignancies, YSTs, choriocarcinoma, embryonal carcinoma, polyembryoma, and mixed GCTs. Finally, gonadoblastoma is a mixed germ cell–sex cord stromal tumor occurring in cases of mixed gonadal dysgenesis with ambiguous genitalia, or 45,X Turner syndrome with Y chromosome material. The histopathology of ovarian GCT types is similar to that of their testicular counterparts. After the third decade of life, ovarian GCTs occur rarely, and carcinomas derived from the coelomic epithelial covering of the ovary are much more common.
Intratubular Germ Cell Neoplasia, Unclassified
IGCNU, also called testicular CIS, is considered the precursor for all invasive testicular GCTs (TGCTs) of postpubertal males except spermatocytic seminoma. According to one source, if left untreated IGCNU has a up to a 50% probability of progressing to invasive TGCT, either seminoma or nonseminoma, within 5 years. Nonseminomas can be composed of embryonal carcinoma (the stem cell component), teratoma (somatic differentiation), and YST and choriocarcinoma (extraembryonic lineages). In IGCNU, germ cells with abundant vacuolated cytoplasm; large, irregular nuclei; and prominent nucleoli are found within the seminiferous tubules, showing similarities to PGCs and early gonocytes. IGCNU is found in 1% of cases of male infertility and in 2% to 4% of cryptorchid testes in adults. In men with a TGCT, the prevalence of IGCNU is about 80% in the ipsilateral testis (range, 63% to 99%], predominantly in men with nonseminomas, and 5% in the contralateral testis. Various markers such as placental alkaline phosphatase (PLAP), c-kit, transcription factor AP-2γ, OCT3/4 (POU5F1), and testis-specific protein Y-encoded (TSPY) have been extensively used for immunohistochemical detection of IGCNU. All these markers are also found in PGCs and/or gonocytes, which supports the model that IGCNU derives from PGCs or gonocytes.
Whether an identifiable precursor lesion exists for GCTs of children is much less clear. In prepubertal children, the incidence of IGCNU appears to be very low, although several cases have been reported. These data must be interpreted with caution, however, because of the difficulty of distinguishing IGCNU in the developing gonad, in which primitive germ cells normally express markers such as OCT3/4 and c-KIT . The precursor lesion for the teratomas and YSTs of neonates and infants is yet to be identified. These pediatric tumors show partially erased imprinting, suggesting that the cell of origin is most likely a germ cell at an earlier development stage than that of IGCNU.
IGCNU has been reported in a high percentage of gonadal biopsy specimens from children with disorders of sexual development (DSDs) who are at risk for the development of germ cell malignancies. These disorders include gonadal dysgenesis, partial androgen insensitivity syndrome (AIS), and less often, complete AIS. Development of these tumors in patients with DSDs is linked to the presence of a specific fragment of the Y chromosome, known as the GBY region. For these patients with an increased risk of germ cell malignancy, prophylactic gonadectomy is often indicated. However, overdiagnosis of IGCNU is possible in this setting because of the common maturation delay of germ cells in patients with DSDs, resulting in retained presence of c-kit -positive germ cells. Therefore careful examination of the distribution pattern of OCT3/4 -positive cells and their position within the seminiferous tubule are essential to making the diagnosis of IGCNU.
Teratoma
Histopathology of Teratoma
Teratomas are tumors composed of multiple tissues that are normally foreign to, and able to proliferate in excess of, the site in which they occur. In the usual definition of teratoma, tissue elements of all three germ layers (endoderm, mesoderm, and ectoderm) are present; however, teratomas also occur in which only one or two of the germ layers are present (referred to as monodermal or bidermal teratomas, respectively). The clinical and biologic behavior of teratomas varies significantly with differing anatomic location, degree of maturity of the tumor tissue, and age of onset. Grossly, teratomas are nodular and heterogeneous with solid and cystic areas depending on the types of differentiated tissue present. Cartilage, bone, hair, and pigmented areas may be recognizable ( Fig. 63-2 ). At the histopathologic level, ectodermal derivatives such as neuroepithelium, skin, and hair are the most common tissues in teratomas of infants and children. However, practically any tissue type can be seen, including muscle, bone, cartilage, and well-differentiated glands ( Fig. 63-3 ).
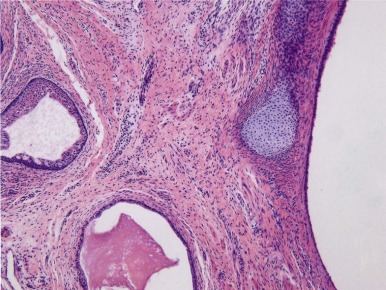
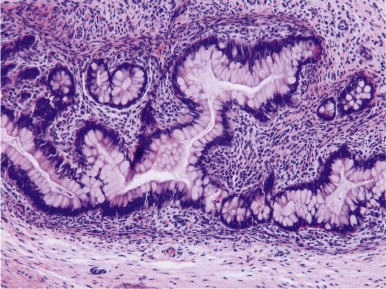
Mature teratomas are typically cystic and contain differentiated adult-type tissue from one or more of the germ layers. The term dermoid cyst refers to the common finding in mature teratomas of tissue resembling the adult epidermis and its appendages. Immature teratomas are those in which embryonal-appearing tissue is present, typically in the form of neuroepithelial rosettes and tubules and often mixed with mature tissue ( Fig. 63-4 ). A loose, myxoid stroma of immature mesenchyme with focal differentiation into osteoid, fat, cartilage, and rhabdomyoblasts may be present. Hypercellularity, increased mitotic index and nuclear atypia can be present. The prognostic significance of these findings is different for ovarian and testicular teratomas, and these are discussed separately in the next sections.
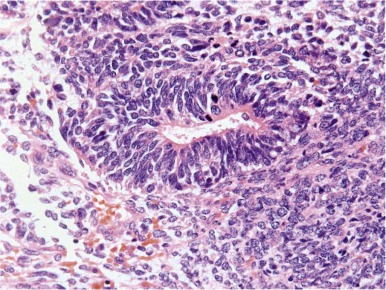
Testicular Teratoma
Testicular Teratoma in Infants and Children
In most series, testicular teratoma is the second most common GCT of the prepubertal testis, representing about 35% of GCTs. However, the actual incidence may be higher, because prepubertal testicular teratomas are uniformly benign and thus may be underreported to tumor registries. Histologically, 85% of prepubertal testicular teratomas are mature, and 15% are immature. Unlike the case of ovarian teratomas, the degree of histologic immaturity of childhood testicular teratoma does not carry prognostic significance. In this age group, pure testicular teratomas lack metastatic potential and have not been found to recur after surgical removal of the tumor. A review of cases of immature teratoma from Pediatric Oncology Group (POG)/Children’s Cancer Study Group (CCG) protocols concluded that the presence of microscopic foci of YST, rather than the grade of immature teratoma, is the only valid predictor of recurrence in pediatric immature teratoma at any site. The benign nature of these tumors has permitted the widespread use of testis-sparing enucleation surgery for prepubertal testicular teratomas. In agreement with the benign clinical behavior of pure teratomas of the prepubertal testis, these tumors display normal karyotypes and normal results from comparative genomic hybridization (CGH). Epidermoid cysts are rare tumors in the prepubertal testis and are most likely monodermal teratomas showing ectodermal differentiation. These tumors, which have a characteristic ultrasonographic appearance, are hormonally inactive and do not produce α-fetoprotein (AFP); as with other teratomas of the prepubertal testis, they are managed conservatively with testis-sparing surgery. In the prepubertal testis, the seminiferous tubules surrounding the teratoma may contain germ cells with atypical features such as enlarged nuclei. However, these features are distinct from the seminoma-like changes of IGCNU, and do not signify malignant transformation of the germ cells.
Testicular Teratoma in Adolescents and Adults
Teratoma occurs as a component of 50% of mixed nonseminomas in the postpubertal testis. Pure teratoma is rare, accounting for only 2% to 3% of postpubertal TGCTs. Testicular teratomas in adolescents and adults may spread, giving rise to teratomatous and nonteratomatous metastases. Furthermore, adult testicular teratomas typically are associated with IGCNU and exhibit characteristic cytogenetic abnormalities such as isochromosome 12p that are also found in seminomas and in other types of nonseminomatous TGCTs. Based on these findings, Ulbright proposed that teratomas in the adult testis arise from a germ cell that has already undergone malignant transformation; thus the histogenesis of adult teratoma is distinct from that of teratomas arising in the ovary or the prepubertal testis. Postpubertal teratomas may contain syncytiotrophoblastic giant cells, and patients with teratoma may have gynecomastia or elevated serum β-human chorionic gonadotropin (β-HCG) or AFP.
Ovarian Teratoma
Teratoma is the most common GCT of the ovary, comprising more than 95% of all ovarian GCTs. Ovarian teratomas differ in important ways from testicular teratomas. Teratomas in the prepubertal testis are uniformly benign. In adolescents and adults, testicular teratomas are nearly always malignant and occur as a component of mixed malignant GCTs (MGCTs), the exception being dermoid cysts of the testis. In the ovary, in contrast, the phenotype of teratomas does not vary as strikingly by age, and ovarian teratomas are usually pure tumors. The great majority of ovarian tumors are mature, benign tumors (often referred to as dermoid cyst or mature cystic teratoma ). Less than 5% of ovarian teratomas are malignant; these are discussed separately in the following sections.
Benign, Mature Ovarian Teratoma
Mature ovarian teratomas are diploid, cytogenetically normal tumors. This contrasts to postpubertal testicular teratomas, which are aneuploid and demonstrate cytogenetic abnormalities such as i12p. Interestingly, when polymorphic molecular markers are assayed, a high percentage of mature ovarian teratomas show a homozygous pattern, indicating that these tumors can arise from a germ cell that has completed meiosis I but not meiosis II. Mature ovarian teratomas are usually cystic, though solid tumors can occur, especially in the first two decades. Other characteristics of mature ovarian teratomas include a well-ordered, “organoid” appearance to the differentiated tissues within the teratoma, and the absence of cytologic atypia. Mature ovarian teratomas are uniformly benign, except in the rare case of postteratomatous “malignant degeneration” of a dermoid cyst.
Malignant Ovarian Teratoma
In children, immature teratoma is the most common type of malignant ovarian teratoma. Also in this category are other tumors that are rare in children, such as monodermal teratomas with malignant elements (e.g., papillary thyroid carcinoma in struma ovarii), dermoid cysts with malignant degeneration, and the rare case of mixed malignant ovarian GCT (MOGCT) with a teratoma component.
Immature Teratoma
The term immature teratoma refers to teratomas containing immature-appearing tissues, principally neuroepithelium. Additionally, foci of mitotically active glia may be present. A grading system has been developed for immature teratomas based on the amount of neuroepithelial tissue present. Mature teratomas containing only fully differentiated tissue are considered grade 0, and immature teratomas range from grade 1 (comprised of less than 10% immature neuroepithelium) to grade 3 (more than 50% immature neuroepithelium present). A two-component grading system (low grade and high grade) has also been proposed and shown to have improved reproducibility. In adult ovarian teratomas, the finding of grade 2 or 3 immature teratoma predicts the likelihood of metastasis and confers a worse prognosis. In children, however, it is not clear that the same relationship of grade to outcome of immature teratomas holds true. A POG study concluded that surgery alone was curative in children and adolescents with stage I immature teratomas of any grade and that chemotherapy should be reserved for cases of relapse.
Immature teratomas may be associated with implants of glial tissue on the peritoneum, known as gliomatosis peritonei . If the implants are composed solely of mature tissues, the presence of gliomatosis peritonei does not “upstage” the patient. Interestingly, data suggest that the implants arise from metaplastic transformation of pluripotent müllerian stem cells in the peritoneum or underlying mesenchyme, rather than representing metastasis of the teratoma tissue.
Monodermal Teratomas
Monodermal teratomas consist largely or exclusively of a single type of tissue, such as thyroid, carcinoid, and neuroectodermal tissues. Monodermal teratomas with neuroectodermal differentiation include indolent types such as ependymoma and poorly differentiated forms such primitive neuroectodermal tumor (PNET), as well as anaplastic tumors resembling glioblastoma. These tumors are rare in children.
Teratoma with Malignant Transformation
Teratoma with malignant transformation makes up 0.2% to 1.4% of mature cystic tumors of the ovary. In these tumors, more commonly seen in older women, somatic-type malignancies are seen among the individual tissues in the teratoma. Squamous cell carcinomas are the most common tumor type, but others including melanoma, adenocarcinoma, small cell carcinoma, sarcomas (including PNET), and malignant glioma may also occur.
Yolk Sac Tumor
Histopathology of Yolk Sac Tumor
YST is the most common malignant GCT in infancy and childhood. The differentiation of the tumor cells is predominantly along endodermal lines and can take the form of both intraembryonic endodermal derivatives, such as primitive gut and liver, and extraembryonic structures such as allantois and yolk sac. There are many synonyms for YST, including orchioblastoma, Teilum’s tumor, and clear cell adenocarcinoma. Although endodermal sinus tumor is commonly in use as a synonym for YST, some consider the term problematic, because the endodermal sinus is not a component of normal human embryogenesis. In macroscopic appearance, YSTs are yellowish, with multiple cysts and areas of hemorrhage, necrosis, and liquefaction. Microscopically, YSTs display a loose, myxoid stroma containing a reticulated pattern of microcystic spaces ( Fig. 63-5 ). The cysts are lined by a flattened, periodic acid-Schiff (PAS)–positive, diastase-resistant epithelium. About 20% of YSTs exhibit characteristic Schiller-Duval bodies (a clustering of cells around a small, central blood vessel) (see Fig. 63-5 ). In addition to the microcystic/reticular pattern, several variant histologies of YST are described, including the solid, polyvesicular, and parietal types (corresponding to primitive endoderm and extraembryonic structures such as the allantois and yolk sac) and the glandular and hepatic types (corresponding to the intraembryonic endodermal derivatives, primitive lung and liver).
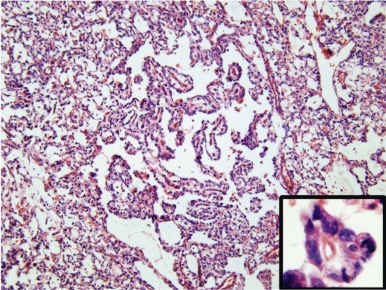
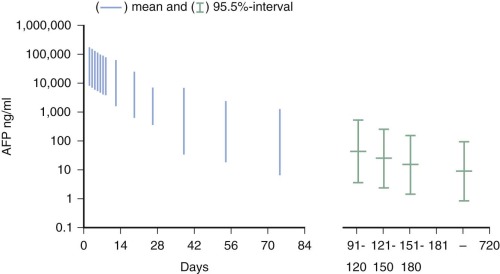
YSTs are typically cytokeratin-positive, which can help differentiate the solid YST variant from germinomas. Alpha-fetoprotein (AFP) expression is characteristic of these tumors, and AFP is a valuable tumor marker. Neonates and infants have high physiologic levels of AFP in serum, which can confound the use of serum AFP levels for diagnosis of YST or for monitoring therapy. Tables of normal serum AFP levels in neonates and infants have been published ( Fig. 63-6 ). In addition, at least 70% of YSTs have detectable AFP expression by immunohistochemical assays.
Testicular Yolk Sac Tumor
Testicular Yolk Sac Tumor in Infants and Children
YST is the most common malignant GCT in infants and children, comprising approximately 65% of prepubertal testicular GCTs. YST is most common in males aged 0 to 2 years and can occur in pure form or as a malignant component of teratoma. Histologically, prepubertal YSTs show the same spectrum of patterns as their adult counterparts: pseudopapillary, reticular, polyvesicular vitelline, and solid, with the pseudopapillary pattern being most common in children. IGCNU is not a feature of childhood YST, and it is unlikely that P53 mutations play a role in pathogenesis. Unlike childhood teratomas, YSTs in children are aneuploid, with characteristic, nonrandom chromosomal abnormalities including gains at 3p and losses at 1p and 6q. Immunohistochemistry detects AFP expression in more than 90% of YSTs, and in most cases the serum AFP level is also elevated.
Testicular Yolk Sac Tumor in Adolescents and Adults
YST in the adolescent and adult testis is extremely rare in the pure form but occurs in approximately 40% of mixed TGCTs. In one series of primary mediastinal GCTs, YST was present in nearly 50% of the tumors. As in childhood YST, hematogenous metastases are common, especially to the liver. The majority of tumors stain positive for AFP, and serum AFP is commonly elevated.
Ovarian Yolk Sac Tumor
YST of the ovary is a clinically aggressive tumor that often presents at advanced stages with metastasis to lymph nodes or peritoneal structures. The tumors are rarely bilateral. The median age at diagnosis is 18 to 19 years. In a series of 26 pediatric patients, reticular type was the most common histology identified.
Seminoma and Dysgerminoma
Histopathology of Seminoma and Dysgerminoma
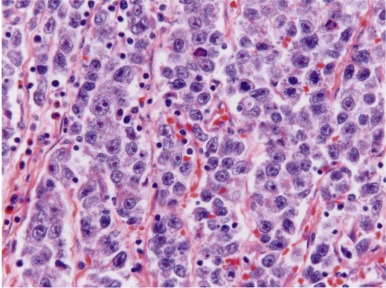
Seminomas are a class of GCTs in which the tumor cells resemble primitive, undifferentiated germ cells, both in morphology and in expression of pluripotency genes such as OCT3/4, NANOG, and STELLAR/DPPA3 . These tumor are referred to as germinomas when arising in extragonadal locations (such as the pineal gland, the mediastinum, or the retroperitoneum) , dysgerminomas when arising in the ovary, and seminomas when arising in the testis. The histology of seminoma is identical regardless of the site in which the tumor occurs. Grossly, these are rounded, nodular tumors, separated into lobules by fibrous bands. Microscopically, seminomas are characterized by a monotonous proliferation of large, uniform cells with large central nuclei and prominent nucleoli ( Fig. 63-7 ). The cytoplasm is clear or granular and often PAS-positive because of high glycogen content. A lymphocytic infiltrate is a typical feature of seminomas.
Seminoma
Seminoma is extremely rare in children. In the postpubertal testis, seminoma can occur in pure form or as a component of mixed GCT. The peak age of onset of seminoma is 34 to 45 years, slightly later than that of nonseminomatous tumors. Several distinct variants of seminoma have been recognized. Up to 7% of classic seminomas contain syncytiotrophoblastic giant cells, and almost 25% of seminomas contain foci that stain positive for β-human chorionic gonadotropin (HCG). These tumors may be accompanied by an elevation in serum β-HCG; however, the presence of syncytiotrophoblasts or elevated β-HCG does not worsen the prognosis. Marked elevations of β-HCG or persistently elevated β-HCG after orchiectomy may indicate the presence of choriocarcinoma. Anaplastic or atypical seminoma is the designation used for seminomas displaying increased mitotic activity, nuclear pleomorphism, and sparse lymphocytic infiltrate on histologic examination. Whether these aggressive features portend a worse prognosis or a higher likelihood of metastasis remains controversial. Whereas some studies have suggested that anaplastic seminomas are clinically more aggressive tumors, others have found no evidence of a worse prognosis when these features are present.
Dysgerminoma
Dysgerminoma is the most common MOGCT of children and adolescents, and makes up one third of MOGCTs. Pathologically, dysgerminoma is the ovarian counterpart of the seminoma of the testis and the germinoma of extragonadal sites. Unlike seminomas of the testis, which are rare in the prepubertal period, dysgerminomas can occur at any age, though the peak incidence is age 15 to 19 years. Also unlike seminomas, which develop from IGCNU, dysgerminomas do not appear to arise from a precursor cell. Dysgerminomas stain positively for OCT4 on immunohistochemistry, which can be useful for distinguishing these tumors from nondysgerminomas. As with seminomas, the majority of dysgerminomas also stain positive for c-KIT.
Embryonal Carcinoma
Histopathology of Embryonal Carcinoma
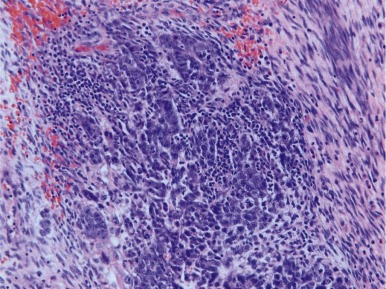
Embryonal carcinoma is very rare in infants but can occur in prepubertal females and adults of both sexes. Embryonal carcinomas commonly have areas of hemorrhage and necrosis and can be locally invasive into the epididymis and spermatic cord in males. At the microscopic level, the tumors consist of large, epithelial-appearing cells that resemble the early embryonic cells of the inner cell mass ( Fig. 63-8 ). The tumor cells may grow in solid, papillary, and reticular patterns, forming many clefts and glandlike structures. The tumors are typically cytokeratin and CD30-positive, but negative for epithelial membrane antigen (EMA), carcinoembryonic antigen, and vimentin. Syncytiotrophoblast cells, when present in the tumor, can produce β-HCG and cause precocious pseudopuberty in premenarchal girls or vaginal bleeding in older women.
Testicular Embryonal Carcinoma in Adolescents and Adults
Embryonal carcinoma is the most common pure nonseminomatous TGCT in adults, representing about 2% to 10% of cases in this age group. Additionally, 80% of mixed malignant TGCTs contain embryonal carcinoma as one component.
Ovarian Embryonal Carcinoma
Embryonal carcinoma occurs in the ovary less commonly than in the testis, accounting for 4% of malignant ovarian tumors. The median age at diagnosis is 14 years, somewhat younger than ovarian YST. Ovarian embryonal carcinoma is more often associated with β-HCG production and with hormonal manifestations such as precocious puberty, amenorrhea, and hirsutism. Owing to the totipotent nature of embryonal carcinoma cells, differentiation into various histologies may be seen including teratomas and syncytiotrophoblastic giant cells resembling choriocarcinoma. Polyembryoma is a rare tumor in which appear multiple embryoid bodies resembling presomite stage embryos. Polyembryoma is considered by some to be an organoid variant of embryonal carcinoma, though others describe it as one component of mixed GCTs.
Choriocarcinoma
Choriocarcinoma is a rare tumor composed of cytotrophoblast, syncytiotrophoblast, and extravillous trophoblast. The tumor can cause anemia and bleeding symptoms, as well as pseudoprecocious puberty because of expression of β-HCG by the tumor cells. Choriocarcinomas are usually large tumors with prominent foci of hemorrhage. The histopathology of choriocarcinoma is a mixture of syncytiotrophoblastic, cytotrophoblastic, and intermediate trophoblastic cells, often mixed in random fashion surrounding areas of hemorrhage and necrosis ( Fig. 63-9 ). Vascular invasion is a common feature of choriocarcinomas.
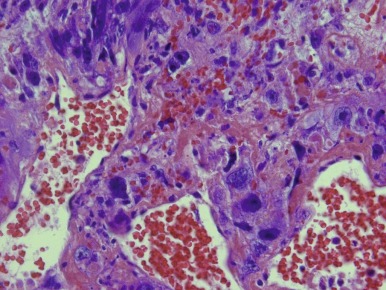
Testicular Choriocarcinoma in Adolescents and Adults
Choriocarcinoma, like YST, is rare in pure form but occurs in approximately 8% of mixed TGCTs in adults.
Ovarian Choriocarcinoma
Primary (nongestational) choriocarcinoma occurs rarely as a pure tumor and more often as a component of mixed ovarian GCT, usually in association with teratoma. Choriocarcinoma in infants may arise as a primary tumor in the lung, liver, brain, kidney, or other location. Treatment of infantile choriocarcinoma is a medical emergency (see “Infantile Choriocarcinoma: A Rare but Rapidly Fatal Condition if Not Recognized.”) Gestational choriocarcinoma arises in the placenta and may metastasize widely in the mother; case reports describe metastasis of gestational choriocarcinoma to the infant. In patients in the reproductive years, deoxyribonucleic acid (DNA) analysis to detect the presence of paternal sequences can be helpful in determining the placental origin of gestational choriocarcinoma.
Mixed Ovarian Germ Cell Tumor
Mixed ovarian GCTs are composed of two or more of the malignant germ cell histologic subtypes. These tumors comprise 8% of malignant GCTs of the ovary in children and adolescents and 20% of ovarian malignant GCTs overall. Mixed GCTs thus represent only about 1% of all ovarian GCTs in contrast to the postpubertal testis, in which one third of GCTs are of the mixed type. Dysgerminoma, YST, and teratoma are the most common germ cell components, with embryonal carcinoma and choriocarcinoma more rarely present.
Pathology of Mixed Germ Cell-Sex Cord Tumors
Gonadoblastoma
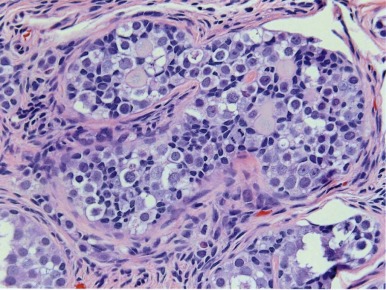
Gonadoblastomas contain mixtures of two cell types: large cells resembling primitive germ cells and smaller cells, similar to immature Sertoli and granulosa cells ( Fig. 63-10 ). It occurs most exclusively in cases of mixed gonadal dysgenesis with ambiguous genitalia, or 45,X Turner syndrome with Y chromosome material. Gonadoblastoma can occur as single or multiple nodules; foci of calcification are a characteristic finding. Microscopically, nests of germ cells within basement membrane material are surrounded by supportive cells (i.e., Sertoli-granulosa cells). The stroma may have elements resembling Leydig or luteinlike cells. The presence of gonadoblastoma substantially increases the risk that a malignant GCT will develop in a dysgenetic gonad. From several studies, the average incidence of GCTs in gonadal biopsy specimens from patients with mixed gonadal dysgenesis is 12%. Gonadoblastoma gives rise most often to dysgerminoma, and less commonly to embryonal carcinoma, teratoma, YST, and choriocarcinoma. The germ cells in dysgenetic gonads abnormally retain their embryonic characteristics, as demonstrated by their marker expression profile, including OCT3/4 and TSPY . These cells give rise to gonadoblastoma in areas of undifferentiated gonadal tissue within the dysgenetic gonad, whereas IGCNU arises in regions with testicular differentiation. * Thus just as IGCNU is the precursor lesion for invasive TGCTs, gonadoblastoma can be considered a precursor lesion to the invasive GCTs of dysgenetic gonads.
* References
Summary of Pathology
The five major histologic subtypes of GCTs are teratoma, YST, germinoma, embryonal carcinoma, and choriocarcinoma. In infants and children, teratoma and YST are the most common GCTs. YSTs occurring at any age are malignant tumors. Teratomas are benign in the prepubertal testis; in the postpubertal testis teratomas occur as one component of nonseminomatous GCTs and are uniformly malignant. Most ovarian teratomas are benign; however, ovarian teratomas with a significant immature component are considered malignant. Germinomas in the ovary are known as dysgerminomas and may occur in children and adults. The testicular counterpart is known as seminoma and occurs exclusively in the postpubertal setting. Extragonadal germinomas can occur in both sexes, typically in the midline of the retroperitoneum or mediastinum or in the pineal gland. The remaining tumor types (embryonal carcinoma and choriocarcinoma) are very rare in children but can occur in the testis or the ovary in adolescents and adults, either as pure tumors or as one component of a mixed GCT.
Epidemiology of Pediatric Germ Cell Tumors
Incidence
Pediatric GCTs account for only a small proportion of all cancer diagnosed in children under age 20, but incidence fluctuates dramatically with age. Teratomas, which are benign, are the most common neoplasm of the newborn. However, there are malignant forms of GCT that arise in early childhood between the ages of 0 and 4 years, particularly extragonadal GCT in girls and TGCT in boys. During adolescence, rates of GCT rise substantially for both males and females. Whereas in children younger than 15 years old, GCTs represent only 3.5% of all cancer diagnoses, during adolescence between ages 15 and 19 years, GCTs represent 16% of the total cancer burden. The incidence of testicular GCTs continues to rise throughout young adulthood, peaking in the third decade, and are the most common malignancy in men between the ages of 15 and 34 years of age. Approximately 8300 cases of testicular cancer are diagnosed among adult males annually.
Extrapolating data from the Surveillance, Epidemiology, and End Results (SEER) cancer registry of childhood cancer collected between 1976 and 2006, it is estimated that in the United States, approximately 850 children and adolescents under the age of 20 are diagnosed with a gonadal or extragonadal malignant GCT each year (see Tables 63-1 and 63-2 ). Incidence of GCT in childhood has a bimodal distribution; there is a peak in diagnosis at birth and up to age 4, and then a second peak after the onset of puberty. Histology varies dramatically with age. YST is the predominant histology in newborns and younger children, whereas tumors that arise in the peripubertal period contain a wider range of histologies, including YST, embryonal carcinoma, and choriocarcinoma, as well as germinomatous tumors (testicular seminoma and ovarian dysgerminoma).
| ICCC Group X, a-e | Description | Total | Males | Females |
|---|---|---|---|---|
| Germ cell trophoblastic and other gonadal tumors | 11.6 | 12 | 11.1 | |
| Xa | Intracranial and intraspinal GCTs | 1.6 | 2.3 | 0.9 |
| Xb | Other and unspecified nongonadal GCTs (This category includes the tumors of infants and young children that originate in the sacrococcygeal region as well as mediastinal tumors primarily developing in older children.) | 1.6 | 1.5 | 1.8 |
| Xc | Gonadal GCTs | 6.7 | 8 | 5.3 |
| Testis | 4.1 | 8 | — | |
| Ovary | 2.6 | 5.3 | ||
| Xd | Gonadal carcinoma | 1.4 | 0.1 | 2.9 |
| Ovary | 1.3 | — | 2.6 | |
| Other | 0.1 | 0.1 | 0.03 | |
| Xe | Other and unspecified malignant gonadal tumors | 0.2 | 0.1 | 0.3 |
| ICCC Group X | ICCC Germ cell Tumor Category | White Male | Black Male | White Female | Black Female |
|---|---|---|---|---|---|
| X a-e | All | 12.3 | 3.2 | 9 | 10.8 |
| Xc | Gonadal GCTs | 9.1 | 1.2 | 4.5 | 5.6 |
| Testis | 9.1 | 1.2 | — | — | |
| Ovary | — | — | 4.5 | 5.6 | |
| Xa,b,d,e | Other than gonadal GCTs | 3.2 | 2 | 4.5 | 5.2 |
The site of GCT varies by gender. Under age 10 years, the overall rates are similar between boys and girls, but the extragonadal site is much more common in girls than boys. Over age 10 years, incidence increases in both males and females, but the magnitude differs. Rate increases sixfold in boys but only 2.5 fold in girls. Site, however, is primarily gonadal in both sexes after puberty.
Incidence rates vary dramatically by race as well. Nonwhite boys have higher rates than white boys under age 10, but over age 10, African-American men have much lower rates than white males. In contrast, the rates of GCT are higher among African-American women at all ages. These striking differences in rates of diagnosis by race persist into adulthood; adult African-American males also have a very low likelihood of being diagnosed with testicular cancer.
Incidence not only varies by race and ethnicity, but the overall incidence of GCT has been steadily increasing over the last several decades. Among pediatric patients, incidence rates between 1975 and 2006 increased among boys ages 10 to 19 years (annual percentage change, 1.2; 95% confidence interval [CI], 0.4 to 2.1) and among girls ages birth to 9 years (annual percentage change, 1.9; 95% CI, 0.3 to 2.5). In England and Wales from 1962 to 1990, investigators examined trends in cancer registry data for testicular cancer in males younger than 15 years of age and reported that on average incidence increased 1.3% per annum over the interval. German investigators also report an increase in the incidence of pediatric GCTs over the last several decades.
An increase in testicular cancer has also been noted in adult males in many of the countries of the developed world since at least 1940. In the most recent analysis of U.S. data, the incidence of testicular GCTs in adult men rose 44% between 1973 and 1998, with a more pronounced rise in seminoma (64%) than in nonseminoma (24%). In a study of 22 European countries, incidence was noted to be increasing between 1% (Norway) to 6% per year (Spain and Slovenia). The fact that similar trends in incidence have been observed in both young children and adults strengthen the hypothesis that it is exposures that occur either prenatally or in early childhood that are responsible for this increase in incidence.
Possible Environmental Etiologies
Given the worldwide increase in testicular cancer of almost 100% in the last 3 decades, and the concomitant rise in pediatric GCTs, there has been significant interest in identifying potential environmental exposures that could be implicated. The parallel rise among both children and young adults has pointed to early life exposures as likely being related to development of GCTs.
One of the primary hypotheses has been that induction of GCTs occurs in the setting of prenatal exposure to high levels of maternal estrogens that could affect gonadal development. Several lines of evidence suggest this is a plausible hypothesis. When diethylstilbestrol (DES) is administered to pregnant mice, it causes maldescent of the testes and testicular hypoplasia. Male offspring of women who took DES during pregnancy have higher rates of cryptorchidism, a known risk factor for TGCT in humans. Maternal use of DES has also been directly associated with increased risk of testicular cancer in some but not all studies.
The fetus is also exposed to high levels of endogenous maternal estrogens during pregnancy (versus exogenous sources such as DES or oral contraceptives). Several conditions of pregnancy are known to be associated with higher levels of serum estrogens and have also been linked to increased risk of GCT, including hyperemesis gravidum and preeclampsia. Testicular cancer is more common among first-born males and twins, especially dizygotic twins, and again, both conditions of pregnancy are known to be associated with higher levels of circulating estrogens. Increased birthweight, which has been associated with higher risk of testicular cancer, is also associated with higher estradiol and estriol levels in both the first and third trimesters of pregnancy. Maternal weight gain is a predictor of higher birthweight and is also associated with higher insulin levels, leading to lower sex hormone–binding globulin levels, which thereby increases levels of bioavailable estrogens.
Furthering the hypothesis of the importance of early life exposures playing a role in testicular carcinogenesis, it has been noted that men born during World War II in Norway, Sweden, and Denmark have a decreased risk of testicular cancer compared with men born before or after the war. Lack of maternal weight gain during wartime may explain in part this temporal association. Rationing during WWII in Norway resulted in a 15% to 20% energy restriction and a resultant decrease in maternal weight in the period from 1941 to 1945. Height, determined in part by childhood nutrition, has been found to be strongly associated with risk of testicular cancer in two case-control studies.
Because GCTs are rare in children, the number of case-control studies conducted among children has been quite limited. The Children’s Cancer Group (CCG) conducted an epidemiologic study that included all children with cancer registered for treatment with CCG between 1982 and 1989. Parents were asked to complete a 22-page self-administered questionnaire. Approximately 50% of those with children who had eligible cancer cases completed the questionnaire; controls were obtained through a random-digit–dialing procedure. Among all the cases, there were 105 cases of children younger 15 years of age with a malignant GCT and 639 controls. In this study, several factors were associated with risk of GCT. Birthweight of more than 4000 g was related to 2.4-fold increase in risk (CI, 1.0 to 5.8) compared with a birthweight of less than 3000 g after adjustment for gestational age. Pregnancy conditions such as nausea and vomiting or hypertension or eclampsia were not associated with risk. An inverse association between maternal smoking and risk was noted; among mothers who smoked more than 15 cigarettes per day, risk of GCT was reduced by 70%. Among mothers who ever reported being exposed to chemicals or solvents or to plastic and resin fumes, an increased risk was observed (odds ratio [OR] 4.6; CI, 1.9 to 11.3); (OR 12.0; CI, 1.9 to 75).
A more in-depth follow-up study that specifically focused on GCTs in children was conducted under the aegis of the Children’s Oncology Group (COG). The study included 278 cases and 423 controls identified through random-digit dialing. Subjects completed a self-administered questionnaire, a telephone interview, and a medical record review of the maternal records before and during the index pregnancy. The study did not confirm a relationship between maternal exposure to exogenous hormones (either DES or oral contraceptives) and risk of pediatric germ tumor. None of the pregnancy-related conditions including birthweight, hyperemesis, preeclampsia, pregnancy-induced hypertension, or maternal weight gain that can alter circulating levels of endogenous estrogen were related to risk. The study also failed to replicate the finding from the prior case-control study in CCG that showed a relationship between maternal smoking and decreased risk of GCT ; neither environmental or occupational exposure to pesticides and other chemicals increased risk of GCT in offspring.
Predisposing Syndromes
Cryptorchidism and Testicular Cancer
Many studies have shown a definitive relationship between cryptorchidism and testicular cancer with relative risks that range from 2.1 to 17.6. Of note, cryptorchidism is three to four times more common in white males than African-American males, which mirrors the difference in incidence by race. The etiology of the relationship between cryptorchidism and testicular cancer remains unclear. It has been suggested that exposure of the testis to higher temperatures in the abdomen as compared to in the scrotum in some way promotes cancer growth. In this case, orchiopexy should minimize risk, especially if it is done before the hormonal stimulation and proliferation of puberty. Others have suggested that the etiology of the undescended testicle and the cancer is the same, and in this case, orchiopexy should not significantly decrease risk. It has been observed that there is an increased risk of testicular cancer in the descended contralateral testicle, which supports this hypothesis for a common etiology. In a case-control study from Kaiser-Permanente of 183 patients with testicular cancer matched to 551 controls, there were 20 men who had had a cryptorchid testes. Interesting, if orchiopexy had been performed successfully by age 11, there was no increase in risk of testicular cancer. However, among men whose cryptorchid testis was not treated until after their eleventh birthday, who had a failed orchiopexy, or who had never had treatment, the odds of testicular cancer were increased 32 fold. Although the study was limited by small numbers, it would suggest that orchiopexy in early childhood before puberty may obviate the risk of subsequent testicular cancer. In the case in which an abdominal testes is discovered after the onset of puberty, an orchiectomy may be the most prudent procedure.
Turners Syndrome and Gonadoblastoma
Turner syndrome, the most common sex-chromosome abnormality in girls (1:2000 live births), is caused by partial or total loss of an X chromosome, resulting in a 45,XO karyotype. Most girls with Turner syndrome have short stature and many have primary amenorrhea. Approximately two thirds of girls have only the remnant of an ovary or “streak gonad.” In these dysgenetic and often nonfunctional ovaries, there is a high risk of developing a gonadoblastoma, particularly in girls who also retain a portion of the Y chromosome. Gonadoblastoma is itself a benign lesion, but some of these tumors undergo malignant degeneration, and therefore gonadectomy is usually recommended, although this recommendation has become more controversial. In a Danish study that examined 114 girls with Turner syndrome, presence of the Y chromosome was documented in 13 girls (12%). In the 10 girls with Y-chromosome material who subsequently underwent gonadectomy, gonadoblastoma was present in only one specimen. Therefore the authors suggest that close follow-up examination by ultrasound may be sufficient for monitoring rather than routine gonadectomy but urged that more prospective data is necessary.
Klinefelter Syndrome
Klinefelter syndrome, a condition present in between 1:500 and 1:1000 males, is caused by an extra X chromosome resulting in the 47, XXY karyotype. Men with Klinefelter syndrome have abnormal testicular development and are usually infertile. Men with Klinefelter syndrome also have a substantially increased risk of developing GCTs, particularly mediastinal GCTs, beginning in early adolescence.
Androgen Insensitivity Syndromes
Mutations either in the androgen receptor itself or its downstream effectors results in phenotypic females with an XY karyotype. The prevalence of this disorder is estimated to be approximately 1:20,000 females. Oftentimes, these patients do not come to medical attention until they undergo evaluation of primary amenorrhea. Patients with complete androgen insensitivity have little to no axillary or pubic hair, no uterus, and serum testosterone levels of a male. Testes are abdominal or inguinal but can also present on rare occasions as labial masses. These cryptorchid testes are at high risk for development of gonadoblastoma and/or more malignant GCTs and gonadectomy is advised.
Molecular Biology of Germ Cell Tumors
Analysis of the GCT genome and transcriptome has begun to yield insight into the pathogenesis of these tumors in recent years. Genome-wide association studies (GWAS) comparing GCT cases with unaffected controls have identified a number of loci linked to GCT susceptibility. Several different types of analyses, described in the following sections, lend support to the idea that prepubertal and postpubertal GCTs arise from different molecular mechanisms. In the coming years, identification of specific gene expression signatures, mutations, copy number aberrations, and other abnormalities in different subclasses of GCTs will undoubtedly help refine risk stratification in newly diagnosed patients, as well as provide new targets for novel therapies, especially in chemotherapy-resistant disease.
The GCT Transcriptome
In the last decade studies have begun to illuminate the messenger ribonucleic acid (mRNA) expression pattern of TGCTs in adult men. These analyses have revealed gene signatures distinguishing GCTs from other types of malignancies and further identified differences in gene expression among different histologic subtypes of GCTs. A major unresolved question of whether pediatric GCTs exhibited the same gene expression patterns was addressed by Palmer, Coleman, and co-workers in an investigation of the pediatric GCT transcriptome, the largest such study to date. This study demonstrated striking differences in the gene expression repertoire of malignant YSTs compared to seminomas, with the former enriched for genes associated with the self-renewing or pluripotent phenotype and the latter enriched for genes associated with differentiation and proliferation. In addition, Palmer et al compared gene expression in childhood GCTs with that in adult TGCT, as described in a study by Korkola et al. The analysis of Palmer and colleagues showed clear differences between the transcriptional profile of pediatric and adult GCTs, even within the same histologic subtype ( Fig. 63-11, A and B ).
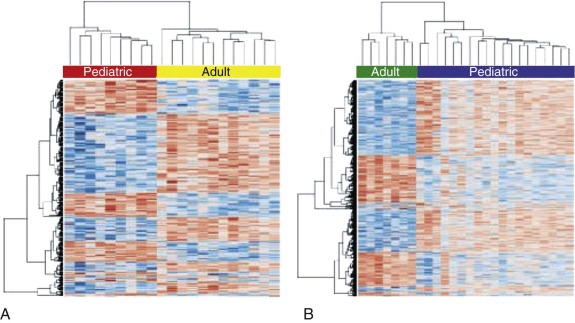
A key additional question is whether particular gene expression patterns are associated with clinical outcome in GCTs, a point addressed by some studies of adult TGCT. A study by Korkola and co-workers identified and validated a prognostic mRNA gene expression signature that defined 5-year overall survival in both a training and validation cohort of nonseminomatous TGCTs. Further work will be required to translate this finding into a robust, clinically useful prediction tool.
Noncoding RNAs
Micro RNAs (miRs) are short, 21-nucleotide, noncoding RNAs that have major roles in regulating gene expression and are often misexpressed in cancer. A seminal study in 2006 identified miR-372 and miR-373 as oncogenes in TGCTs—one of the first demonstrations that miRs play a role in human cancer. In childhood GCTs, Palmer and co-workers demonstrated that relative to normal gonadal tissue, MGCTs overexpress miRs from the miR-371~373 and miR-302 clusters, regardless of histologic subtype, patient age, or gonadal vs. extragonadal origin. This study also demonstrated that expression of these miRs causes specific downregulation of their target miRs in GCTs. In a subsequent study, Murray et al. further defined miRs with differential expression that distinguished YSTs from germinomatous GCTs and identified a core set of transcription factors that may be regulated by these miRNAs and contribute to the specific cellular phenotypes of different subtypes of GCT.
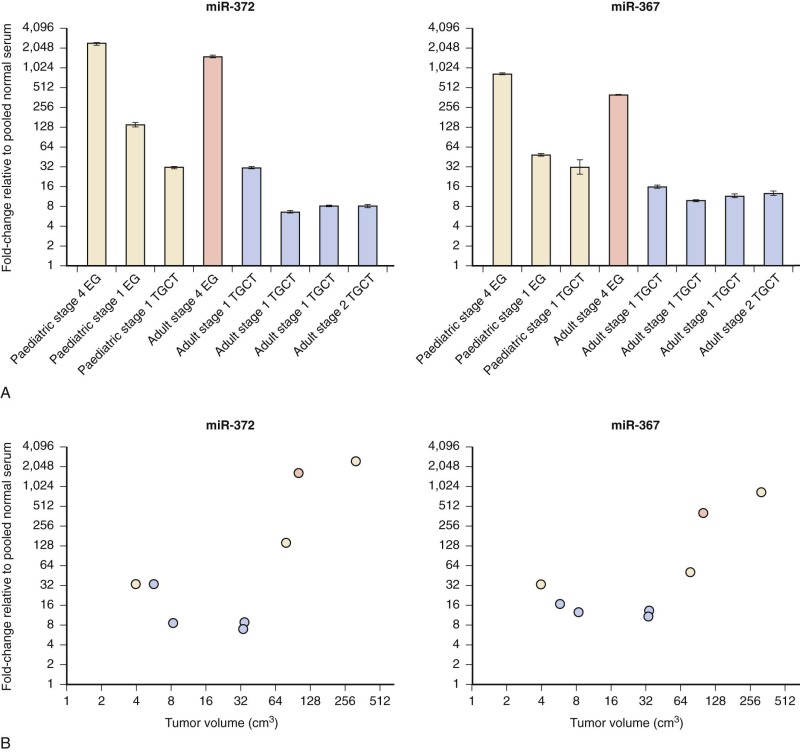
Micro RNAs have been shown to be shed from tumor cells into the circulation. The translational significance of the finding that miR-371~373 and miR-302 family members are universally overexpressed in GCTs became clear with the demonstration that these same miRs were present at elevated levels in the serum of a child with YST at diagnosis and fell in parallel with declining AFP levels during treatment. Further studies have confirmed that serum levels of these miRs are increased in patients with GCT at diagnosis, regardless of age group, tumor histology, or location ( Fig. 63-12 ). This is a particularly important finding, since many patients with GCT do not manifest elevation of serum tumor markers such as AFP, HCG, or lactate dehydrogenase (LDH). Serum miRs may thus represent sensitive and specific new tumor markers for MGCTs.
Chromosomal Constitution and Copy Number Aberrations
The analysis of tumor karyotypes and genomic copy number aberrations (CNAs) in GCTs provides some of the most important evidence for distinct pathogenic mechanisms of GCT in children compared with adolescents and adults. In addition, the gain or loss of specific chromosomal regions may provide important clues as to the molecular mechanisms of germ-cell tumorigenesis.
Adolescent and adult malignant TGCTs are universally aneuploid, and polyploidization of germ cells has been emphasized as an early event in testicular germ-cell tumorigenesis. IGCNU and seminoma cells are hypertriploid, whereas nonseminomas of all histologic types are hypotriploid. TGCTs consistently exhibit biallelic expression of imprinted genes. Erasure of parental imprinting occurs in mammalian embryonic germ cells, implying the origin of TGCTs from an early germline cell.
The most common cytogenetic aberrations in TGCTs are relative loss of all or parts of chromosomes 4, 5, 11, 13, 18, and Y and gain of material from chromosomes 7, 8, 12, and X. Of these, the most consistent chromosomal aberration in adolescent/adult malignant GCTs is the overrepresentation of chromosome 12p, which is found in all histologic subtypes and primary sites (ovarian, testicular, and extragonadal). A characteristic isochromosome 12p (i[12p]) can be detected cytogenetically in 80% of cases; in other cases, an increased copy number of 12p sequences can be detected on derivative chromosomes. Along with tumor markers such as β-HCG, the detection of i(12p) can identify a germ-cell origin in cases of carcinoma of unknown primary. Further analysis has identified a particular amplicon, 12p11-12p12.1, that is present in many TGCTs. Candidate genes in the 12p region include the oncogenes KRAS and CCND2 , as well as the stem cell–specific genes NANOG and STELLAR/DPPA3 ; however, the precise contribution of 12p amplification to the pathogenesis of GCTs remains unclear. * 12p amplification appears to be a late event in GCT pathogenesis, because it is not present in the precursor lesion, IGCNU.
* References
Molecular genetic analysis of teratomas further supports the hypothesis of different histogenetic origins of different types of teratomas. The malignant teratoma component of mixed ovarian or TGCTs uniformly exhibits the same genomic aberrations as the other malignant components of the tumor, including i(12)p. In contrast, mature, cystic ovarian teratomas and dermoid and epidermoid cysts of the testis exhibit normal karyotypes. † Based on these observations, Ulbright has proposed that mature ovarian teratomas develop directly from transformed germ cells, whereas the post-pubertal testicular teratomas develop through a malignant IGCNU stage, as do the other malignant post-pubertal TGCTs.† References
A recent study of genome-wide copy number variation (CNV) in seminomas using high-resolution single-nucleotide polymorphism (SNP) arrays confirmed many of these reported findings and identified novel CNVs, including both amplifications and homozygous deletions. Several genes identified in regions of recurrent CNV are potential candidate seminoma susceptibility genes, including FAT3, EDNRB1, and RASSF8 .Genomic aberrations seen in MGCTs of infants and children are generally distinct from those occurring in postpubertal tumors. Pediatric GCTs show only partial erasure of parental imprinting, indicating that prepubertal GCTs likely arise from an earlier stage of embryonic germ cell development than do the postpubertal tumors. ‡ A similar pattern of imprinting is seen in gonadal and extragonadal pediatric GCTs, suggesting that gonadal and nongonadal tumors share a common pathogenesis and cell of origin.
‡ References
In analyses of chromosomal structure and copy number imbalances, differences between the pediatric and adolescent/adult GCTs are again consistently found. In the prepubertal period, pure teratomas of the testis or of extragonadal sites nearly always exhibit a normal profile in genomic analyses, including classical cytogenetics, fluorescence in situ hybridization, loss-of-heterozygosity analysis, and array CGH. § These data contrast sharply with the universally abnormal cytogenetic profile of postpubertal teratomas arising as a component of a mixed malignant GCT. In adolescents and adult GCTs, only mature teratomas of the ovary and dermoid and epidermoid cysts of the testis exhibit normal cytogenetics, suggesting that these tumors may share a common pathogenesis with the prepubertal teratomas.§ References
In children under the age of 5 years, YST is the most common malignant GCT of the testis and of extragonadal sites. Unlike teratomas, cytogenetic and other genomic aberrations are consistently reported in analyses of YSTs in infants and children. The most common imbalances reported are gains at chromosomes 1q and 20q and losses at chromosome 1p and chromosome 6q. ¶ More recently, the advent of higher-resolution techniques such as array-based CGH has further defined chromosomal aberrations in this group of tumors. Veltman et al reported the array CGH profiles of a series of 24 GCTs from patients aged younger than 5 years, including 16 teratomas and eight YSTs. Most of the teratomas had normal CGH profiles, with two teratomas exhibiting loss of chromosome 20p as the only recurrent change. In contrast, all of the YSTs had abnormal profiles. The recurrent changes, seen in at least three of the eight YSTs, included gains of chromosomes 1q (1q32-1qter), 3p (3p21-pter), and 20q (20q13) and loss of chromosomes 1p (1p35-pter), 6q (6q24-qter), and 18q (18q21-qter). In the Veltman et al study, only one tumor from a child aged younger than 5 years exhibited gain of chromosome 12p, whereas five of seven cases from children older than 5 years showed gains at chromosome 12 p. Most of these tumors occurred in the ovary, supporting the hypothesis that ovarian and testicular MGCTs have a common pathogenesis, at least in older children and adults. Another study from the U.K. Children’s Cancer and Leukaemia Group examined the metaphase CGH profile of 34 malignant GCTs in children aged younger than 16 years. This study supported the previous reports of an increased frequency of loss of chromosomes 1p and 6q and gain of chromosome 3p in YSTs. Intriguingly, 4 of 14 MGCTs from children younger than 5 years of age demonstrated gain of 12p, including in two cases gain of the 12p11 locus that is strongly associated with adolescent and adult MGCTs. Therefore childhood and adolescent/adult MGCTs may share more pathogenic mechanisms in common than has been previously appreciated based on the analysis of a relatively small number of cases.¶ References
Loss of chromosomes 1p and 6q correlates with loss-of-heterozygosity analysis indicating true allelic loss in these regions in pediatric GCTs. However, the most common chromosomal aberrations of pediatric GCTs 1p−, 1q+, 6q−, and 20q+ are not highly specific for GCTs but are seen in other cancers of childhood, including neuroblastoma and Wilms tumor, as well as carcinomas. Deletion of chromosome 1p is associated with MYCN amplification and poor prognosis in neuroblastoma, which has led to the speculation that one or more genes in this interval may act as a tumor-suppressor in neuroblastoma. Several candidate genes have been proposed, including CHD5, TNFRSF25, CAMTA1, and AJAP1. CHD5 was shown to act as a tumor-suppressor in vivo. Whether these or other genes in the 1p and 6q deleted regions play a pathogenic role in pediatric MGCTs is not known.Genome-Wide Association Studies
A number of genome-wide studies have used SNP arrays to analyze the constitutional (germline) DNA of men with TGCT, resulting in the identification of a large number of novel GCT susceptibility loci. In 2009 two groups reported strong linkage of GCT susceptibility to loci on chromosomes 5, 6, and 12, suggesting roles for SPRY4 and KITLG , both components of receptor tyrosine kinase signaling, as well as for the proapoptotic BAK1 gene. A follow-up study by Turnbull and co-workers identified additional susceptibility loci near the DMRT1 , TERT, and ATF7IP genes. The studies have subsequently been replicated in other patient populations. More recently, further GWAS studies have identified new candidate GCT susceptibility loci, including genes with potential roles in telomere regulation, such as PITX1 , or germ-cell development, such as TEX14, RAD51C, PRDM14, and DAZL .
The Role of Developmental Signaling Pathways in Germ Cell Tumor Pathogenesis
Compared with many other solid malignancies, relatively few somatic mutations have been described in GCTs. The most commonly reported mutated gene is KIT , a tyrosine kinase growth factor receptor that plays important roles in germ cell development. Mutations have also been reported in NRAS and KRAS , signaling components of the mitogen-activated protein (MAP) kinase pathway that act downstream of KIT . Interestingly, somatic mutations in BRAF , another MAP kinase pathway member, have been associated with cisplatin resistance in adult TGCTs ; however, BRAF mutations appear to be rare in pediatric GCTs.
Large-scale genomic profiling projects, now underway in several centers, will be required to fully delineate the mutational landscape of MGCTs. Other mechanisms impairing normal gene expression in GCTs such as CNV and aberrant miR expression are being explored (described previously), as is differential methylation. Epigenetic modulation of gene expression through promoter DNA methylation is commonly found in adult and pediatric GCTs. Smiraglia and co-workers first reported that seminomas are globally hypomethylated compared to nonseminomas, whereas Netto et al. extended these observations to show that IGCNU cells, like seminomas, appear globally hypomethylated. Other groups have examined the potential contributions of differential methylation to TGCT susceptibility or cisplatin resistance. Two recent studies examined global DNA methylation patterns specifically in pediatric GCTs. Jeyapalan and co-workers found that pediatric YSTs exhibited a “methylator” phenotype, perhaps attributable to higher expression of DNA methyltransferase 3B in these tumors. Among the genes predicted to be silenced were several associated with caspase-8–dependent apoptosis. Amatruda et al. found a similar pattern of differential methylation comparing YSTs to seminomas and further identified effects on the expression of genes associated with stem cell pluripotency and Wnt/β-catenin signaling.
In addition to these efforts, many studies have focused on understanding how aberrant control of normal developmental pathways may play a role in GCT pathogenesis. Fritsch and co-workers showed that pediatric YSTs and teratomas express active Wnt/β-catenin signaling and Wnt target gene expression. Pediatric YSTs express high levels of BMP2 , a component of the BMP signaling pathway, and expression of BMP7 is associated with poor outcome in TGCTs. The importance of BMP was further revealed in the analysis of zebrafish bearing inactivating mutations in a cell-surface BMP receptor, which develop GCTs strongly resembling human seminomas. Thus the state of BMP signaling distinguishes undifferentiated tumors such as seminomas from the more differentiated nonseminomas.
Another key developmental pathway concerns the pluripotency gene LIN28 , which is universally expressed in GCTs. Whereas LIN28 expression is critical for normal embryonic development, prolonged or aberrant LIN28 expression in germ cells may be pathogenic, owing to LIN28 ‘s ability to block maturation of the let-7 family of tumor-suppressing miRs. Let-7 normally regulates a number of genes associated with proliferation such as MYCN , RAS , AURKB, and CCNF , and Murray et al. have shown that LIN28 expression drives the upregulation of many of these oncogenes in GCT cells.
Although the above studies have begun to yield important insight into the mechanisms by which perturbations in the Wnt, BMP and LIN28 /let-7 pathways may contribute to GCT pathogenesis, strategies to target these defects are still at fairly early stages of development. There may potentially be more immediate translational impact from the finding that GCTs express cell-surface proteins that have been successfully targeted by monoclonal antibodies in other solid tumors, including ERBB2/Her-2 , CD30 , and epithelial cell adhesion molecule (EpCAM).
Diagnosis and Staging of Pediatric Germ Cell Tumors
In this section, the clinical presentation of GCTs, evaluation of the patient for metastatic disease, and the use and interpretation of the tumor markers in the diagnosis and staging of GCTs will be discussed. The surgical approach, for purpose of both diagnosis and staging, will also be delineated.
Clinical Presentation of Pediatric Germ Cell Tumors
The clinical presentation of a pediatric GCT is determined by the site of origin. Site of origin varies by age. Gonadal tumors, particularly testicular tumors, do present during infancy but are more common after the onset of puberty. The site of the extragonadal GCT also varies as a function of age at diagnosis. Sacrococcygeal tumors, much more common in females than males, present either at birth (typically mature/immature teratomas) or between 6 months and 4 years of life (typically MGCT with a large component of YST). Mediastinal tumors are rare in infancy; incidence begins to rise at onset of puberty, particularly among males. Site-specific features of the presentation are discussed in the sections that follow.
Presentation of Testicular Germ Cell Tumors
Testicular GCTs most often present with painless generalized swelling of the scrotum. Gynecomastia and infertility are rarely present, though subfertile and infertile men do appear to have an increased risk of developing TGCT. Local extension to the spermatic cord and the epididymis may occur, and the presenting symptoms can be mistaken for epididymitis. Symptoms of metastasis, including back or abdominal pain, cough or dyspnea, are present at diagnosis in about 10% of adult patients.
An initial testicular ultrasound that shows the lesion to be intratesticular with areas of microcalcification is often helpful in clarifying the differential diagnosis, which includes lymphoma and paratesticular rhabdomyosarcoma. Eighty-five percent of children younger than age 4 have a tumor that is confined to the testes (stage I), as compared with only 35% of adults.
Choriocarcinoma is the most aggressive histologic subtype of testicular GCT, with a propensity to hematogenous metastasis. Patients may have an occult primary tumor and few or no testicular complaints but rather symptoms caused by hemorrhage of visceral metastases, such as hemoptysis or neurologic symptoms resulting from brain metastases. Because of the risk of hemorrhage, the suspected diagnosis of metastatic choriocarcinoma is a medical emergency that requires prompt initiation of treatment, often without confirmatory biopsy and just on the presumption of the diagnosis given the presenting signs, symptoms, radiologic appearance, and markedly elevated β-HCG. However, it has been hypothesized that chemotherapy may actually precipitate hemorrhage, and therefore patients need very careful monitoring; some have advocated initiation of chemotherapy in an intensive care–unit (ICU) setting. With the extreme elevation of the serum β-HCG that can be seen in choriocarcinoma, occasional patients manifest thyrotoxicosis or gynecomastia resulting from the cross-reactivity of β-HCG with the thyroid and luteinizing hormone receptors.
Presentation of Ovarian Germ Cell Tumors
Ovarian GCTs typically present with abdominal pain and a palpable pelvic-abdominal mass (85%). Symptoms of acute abdomen occur in about 10% to 25% of patients and are usually caused by rupture, hemorrhage, or torsion of the ovarian tumor that requires immediate surgical exploration. Other less common presenting symptoms include abdominal distension (35%), fever (10%), and vaginal bleeding (10%). Rarely, patients can experience precocious puberty resulting from the production of β-HCG by the tumor. Not uncommonly, a young girl with an abdominal/pelvic mass and and an elevated β-HCG is assumed to be pregnant. This assumption is especially distressing for young females who are peripubertal and have not yet have initiated sexual intercourse.
If the diagnosis of dysgerminoma is made, it is important to recognize that 10% to 15% of cases are bilateral; therefore the finding of dysgerminoma on frozen section during surgical exploration of an ovarian mass generally warrants close inspection and sometimes biopsy of the contralateral ovary. Most dysgerminomas present as stage I (disease confined to the ovary), but lymphatic metastasis of these tumors to pelvic, paraaortic, mediastinal, and supraclavicular lymph nodes can occur. Five percent of dysgerminomas arise in dysgenetic gonads of girls with 46XY (Sweyer syndrome or testicular feminization) or who have 45X/46XY mosaicism. For this reason, karyotyping should be considered for all girls with a prepubertal pelvic mass. If dysgenetic gonads and/or the presence of Y-chromosome material are detected, removal of both of the gonads is indicated because of the 25% to 50% risk of developing malignancy such as a gonadoblastoma or dysgerminoma in the contralateral ovary. See “ Epidemiology of Pediatric Germ Cell Tumors ” for more information.
Presentation of Sacrococcygeal Germ Cell Tumor
Sacrococcygeal teratomas (SCTs) occur predominately in females. In Altman’s classic 1974 review of 405 clinical cases, 74% occurred in females. However, even though SCTs are much more common in females, rate of malignancy does not vary by gender. Of note, 18% of patients with SCT had associated congenital anomalies. The most prevalent defects were in the musculoskeletal system, but other organ systems that were involved included renal, cardiovascular, gastrointestinal, and central nervous systems. More than half of the patients were diagnosed on the first day of life, but approximately 10% were diagnosed after 1 year of age.
Altman classified sacrococcygeal tumors by the degree of internalization vs. externalization of the tumor ( Fig. 63-13 ). An “Altman type I” SCT is a tumor that is entirely external; an Altman type IV SCT is entirely internal. Altman was the first to note the correlation between rate of malignancy, age at diagnosis, and degree of internalization. At younger than 2 months of age, 7% of girls and 10% of boys had malignant tumors, whereas after 2 months of age, more than half of the tumors were malignant (48% of SCT in girls and 67% of SCT in boys).
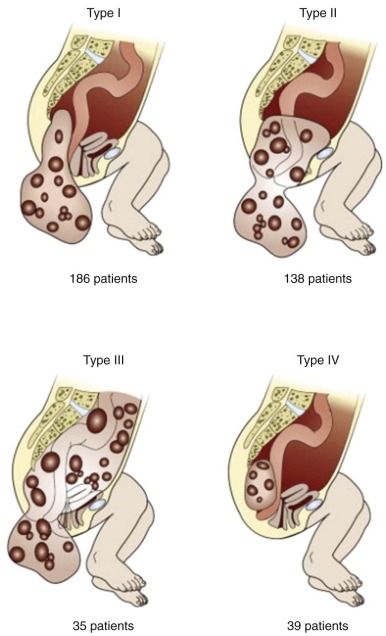
Most neonatal SCTs have a significant external component (Altman type I or II) and can be completely excised in the neonatal period. Over 90% are benign lesions (either mature or immature teratoma) that do not require further therapy. Recurrence rates after resection of neonatal mature or immature teratoma have been estimated to range from 4% to 21% with a malignancy rate of 50% to 70%. The reason that a fraction of neonatal tumors do recur could either be because of oversight of a malignant element during the initial pathologic review, such as a microscopic foci of YST, or because malignant transformation of a small benign remnant left in situ after the initial procedure. Presacral tumors that are predominately internal present later in infancy through age 4 years. Usual presenting symptoms of an internal lesion are either constipation or a buttock/abdominal mass; probability is quite high that these lesions are malignant.
Presentation of Mediastinal Germ Cell Tumors
In a series of patients with mediastinal GCTs registered on the German MAKEI protocols, chest pain or respiratory symptoms such as cough or dyspnea were the most common presenting clinical symptoms. Other presenting clinical scenarios included discovery of the mass as an incidental finding on fetal ultrasound or during a workup for pneumonia that did not resolve with standard antibiotics. In the MAKEI experience, all mediastinal tumors that presented at younger than 1 year of age (n = 9) were either mature or immature teratomas. Mediastinal tumors presenting after infancy but before puberty were either pure YSTs or had predominant yolk sac components. Embryonal carcinoma was more prevalent after the age of 10 years. Mediastinal seminoma was not diagnosed before age 10 years.
Presentation of Extragonadal Germ Cell Tumor at Other Sites
Extragonadal GCTs are the most common fetal and neonatal neoplasm; perinatal teratomas present at wide array of sites. In a review of all perinatal GCTs reported in the literature from 1965 through 2004, the author identified 534 cases. The sacrococcygeal site is the most common (40%), but perinatal GCTs have also been reported in the cervical area (30%), in the oropharynx and nasopharynx (8%), and in other sites including cardiac (7.5%), gastric (2.6%), orbital (2.4%), facial (1.5%), mediastinal (2.6%), and placental (1.5%) locations. In addition, there were 17 teratomas reported in other sites, including tongue, tonsil, liver, retroperitoneum, ileum, mesentery, vulva, and the anorectal area. In addition, 25 cases of fetus in fetu were reported. Most common reasons for diagnosis included detection of a mass on prenatal ultrasound, polyhydramnios, or respiratory distress. Other symptoms were specific to site; for instance, pericardial effusion and tamponade were the most common signs in neonates with intracardiac teratomas. The vast majority of perinatal GCTs are benign, either mature or immature teratomas. Overall the incidence of YST within the neonatal teratoma was 5.8%; the highest rate of YST (10%) was documented in SCTs. Teratomas detected antenatally had three times the mortality rates as those diagnosed postnatally. Fetal survival rate was 53%; neonatal survival rate was 85%. Cause of death was most commonly either because of prematurity or to hydrops. Survival rate was only 7% among those patients whose tumors were diagnosed as a result of fetal distress before 30 weeks of gestation.
Evaluation for Metastatic Disease
The current system for staging pediatric GCTs in use by the COG is shown in Table 63-3 . All patients suspected of having a GCT should have a radiologic evaluation of the extent of the disease including either an ultrasound (if testes is the primary site), computed tomography (CT), or magnetic resonance imaging (MRI) of the primary site; abdominal/pelvic CT if the tumor is likely to drain to the lymph nodes of the retroperitoneum and abdomen; and a chest CT. Radiologic staging is preferably obtained preoperatively so that the clinician does not have to differentiate between malignant involvement and postoperative changes (i.e., atelectasis on the chest CT or postoperative lymph node enlargement after surgery). GCTs can also metastasize to brain and bone. Although bone and brain metastases are generally considered quite rare, German investigators reported that up to 10% of patients have bone metastasis, and that incidence was as high as 26% among stage IV patients. On the last combined U.S. intergroup GCT study, 6% of patients overall had bone metastases, but the incidence increased among stage IV patients, of whom 13% had bone metastases at diagnosis. Brain metastases are present in about 4% of adults with GCT. In a review of the St. Jude experience in 206 cases of pediatric GCT from 1962 to 2002, 16 patients in the series had brain metastases at some point during treatment. Only two patients (1%) had brain metastases at diagnosis. Twelve patients were diagnosed at relapse, and 2 patients were discovered to have brain metastases at autopsy. Most patients with brain metastases had clinical symptoms (12/16) and also had concurrent pulmonary metastases (14/16). The data may overestimate the incidence of brain metastases, however, because 11/16 were diagnosed before 1982 when cisplatin-based chemotherapy was not routine. Risk factors for the development of brain metastases in the St. Jude’s study included extragonadal site ( P = .013), advanced stage ( P = .02) and choriocarcinoma ( P < .001). In comparison, in a more recent cohort of the patients treated from 1990 through 1996 in the U.S. pediatric intergroup study, only three out of 299 patients were diagnosed with brain metastases; two of these patients were stage IV at diagnosis. All patients had other sites of metastatic disease; the histology of all three patients included choriocarcinoma. Two of three patients are alive and well; one was treated with PEB alone and one patient received bleomycin, etoposide, and cisplatin (PEB) plus x-ray therapy (XRT). Therefore a bone scan should be obtained in all patients with advanced stage disease (stage III or IV), but head CT or brain MRI are recommended only for patients with stage IV disease, particularly in patients who have choriocarcinoma.
| Testicular | |
| I | Limited to testis, completely resected by high inguinal orchiectomy; no clinical, radiographic or histologic evidence of disease beyond the testis; tumor markers normal after appropriate half-life decline: patients with normal or unknown markers at diagnosis must have negative ipsilateral retroperitoneal lymph node sampling to confirm stage I disease. |
| II | Transscrotal orchiectomy; microscopic disease in scrotum or high in spermatic cord (<5 cm from proximal end): retroperitoneal lymph node involvement (<2 cm) and/or increased tumor markers after appropriate half-life decline |
| III | Tumor-positive retroperitoneal lymph node(s) >2 cm diameter: no visceral or extra abdominal involvement |
| IV | Distant metastases that may include liver |
| Ovarian | |
| I | Limited to ovary, peritoneal washings negative for malignant cells; no clinical, radiologic, or histologic evidence of disease beyond the ovaries (gliomatosis peritonei did not result in upstaging): tumor markers negative after appropriate half-life decline |
| II | Microscopic residual or positive lymph nodes (<2 cm); peritoneal washings negative for malignant cells (gliomatosis peritonei did not result in upstaging); tumor markers positive or negative |
| III | Gross residual or biopsy only; tumor-positive lymph node(s) >2 cm diameter; contiguous visceral involvement (omentum, intestine, bladder); peritoneal washings positive for malignant cells |
| IV | Distant metastases that may include liver |
| Extragonadal | |
| I | Complete resection at any site; coccygectomy included as management for sacrococcygeal site; negative tumor margins |
| II | Microscopic residual; lymph nodes negative |
| III | Gross residual or biopsy only; regional lymph nodes negative or positive |
| IV | Distant metastases that may include liver |
The use of fluorodeoxyglucose F 18 positron emission tomography (PET) has not been systematically evaluated in pediatrics, but its use is becoming more routine among adult patients with GCTs in the setting of a residual mass postchemotherapy. In a German study that compared the sensitivity and specificity of three different modalities for the evaluation of the residual mass, PET vs. CT vs. tumor markers, the results were as follows: PET, 59% sensitivity and 92% specificity; CT, 55% sensitivity and 86% specificity; and tumor markers, 42% sensitivity and 100% specificity. The positive and negative predictive values for PET were 91% and 62%, respectively. Teratomas are not PET-avid. The authors concluded that PET did not add to the preoperative inference in the setting of a mass in which viable tumor was suspected by CT and tumor marker, but it was a useful adjunct in the setting in which tumor markers had normalized or the patient had marker-negative disease at diagnosis. PET-negative masses also require resection because of the possibility of residual teratoma that is PET-negative.
Tumor Markers
Specific subtypes of GCTs secrete proteins that can be used as markers of tumor presence. The pattern of these tumor markers and degree of elevation provide an indication of the likely histology ( Table 63-4 ). AFP is a glycoprotein that is synthesized by the fetal liver and yolk sac. AFP is elevated in patients with YST, although low levels of AFP (generally <100 IU) are often observed in immature teratoma (perhaps because of occult microscopic foci of YST within the tumor). When an elevated AFP level is detected, one must consider other reasons for an elevated AFP in the differential diagnosis, including synthesis by a liver tumor, such as a hepatoma or hepatoblastoma, or other disease states such as hypothyroidism, folate deficiencies, autoimmune disorders, acquired immunodeficiency syndrome (AIDS), congenital heart defects, cystic fibrosis, and platelet aggregation disorders. β-HCG is a peptide hormone produced in pregnancy, that is made by the embryo soon after conception and later by the syncytiotrophoblast (part of the placenta). Its role is to prevent the disintegration of the corpus luteum of the ovary and thereby maintain progesterone production that is critical for a pregnancy in humans. In tumors that originate in extraembryonic tissues, like choriocarcinoma, β-HCG can be significantly elevated. β-HCG is also sometimes mildly elevated in seminoma/dysgerminoma (<50 IU/L).
Interpretation of AFP levels must incorporate knowledge of age-related norms (see Fig. 63-6 ). AFP is elevated in all infants at birth because there of continued synthesis of AFP by the fetal liver. Over the course of the first 2 years of life, AFP declines in normal infants as the synthesis in the liver ceases. To establish normal values for infants, Blohm et al analyzed data on 414 full-term infants, 90 preterm infants, and 259 children up to age 2 years from the University Hospital Düsseldorf (Germany). Using regression analysis, the authors estimated that the half-life of AFP increases with age over the first 2 years of life. From birth to day 28, the half-life is estimated to be 5.1 days. The half-life of AFP increases thereafter to 14 days at 1 to 2 months of age, 28 days from 2 to 4 months of age, and 42 days from 4 to 6 months of age. The increase in the half-life of AFP with age also results in a “shouldering” of the AFP curve during the first 2 years of life. Not all children have adult levels of AFP by 2 years of age. In the Blohm study, the mean AFP at age 2 was 8 ng/mL; however, the 95% range extended between 0.8 and 87 ng/mL. Similar results were reported in an earlier and smaller study by Wu et al. Values of AFP obtained in children under age 2 ought to be plotted on the nomogram developed by Blohm et al to determine whether the value falls within the normal range.
Tumor markers may also be prognostic. In adult males with testicular cancer, the prognostic significance of postoperative tumors markers is well established. An international consortium pooled data on over 5000 men with testicular cancer and used multivariate analysis to investigate prognostic factors that created the International Germ Cell Consensus Classification (IGCCC), which divided patients into good, intermediate, and poor risk groups based on the extent of elevation of tumor markers, the presence of nonpulmonary visceral metastases or a nontesticular primary site of disease. Although elevated tumor markers are one of the primary variables that determine risk group assignment in adult men, evaluation of the prognostic significance of tumor markers in pediatrics has been limited. In adult males, the failure of tumor markers to decline appropriately among poor-risk patients has also been shown to be a poor prognostic feature. Further discussion can be found in “Prognostic Factors at Diagnosis of Pediatric Germ Cell Tumors.”
Tumor markers are also used prospectively to differentiate between stage I and II tumors. For patients with gonadal GCTs who have had all disease completely removed surgically and in whom no evidence of further disease is noted on pathologic or radiologic staging evaluations a watch-and-wait strategy is often recommended. However, if tumor markers fail to decline according to the expected half-life, the patient by definition has occult metastatic disease. The patient should be upstaged to stage II and therapy commenced.
There is one situation in which tumor markers may increase and not be cause for alarm. During the first cycle of chemotherapy, it has been observed that tumor markers can increase transiently. However, levels should have declined to the expected level by the start of the second cycle of chemotherapy.
Surgery and Germ Cell Tumor
Surgery is an integral part of the management of GCTs. For benign tumors, complete excision is curative. For malignant tumors, proper staging and appropriate timing of excision are important for optimal outcome. At all sites, primary excision should be considered if the tumor can be safely removed without significant risk of morbidity. For tumors that pose risk to adjacent structures or expectation of incomplete excision, biopsy should be performed for diagnosis and neoadjuvant chemotherapy should be given. Reduction in tumor size allows greater expectation of safe and complete excision. Evidence is mounting that delayed resection of extragonadal tumors does not just decrease morbidity but also more importantly may increase survival, because of the ability to achieve complete resection at the time of the delayed surgery. In the case of expected malignancy, a child should be referred to a center with surgeons with experience in oncologic surgery, preferably a site that participates in COG clinical trials so that the proper staging can be done to allow for trial entry.
Surgical Approach to Sacrococcygeal Tumors
Over 90% of neonatal sacrococcygeal tumors are benign (mature or immature teratoma), and require complete excision at diagnosis should be attempted without harming adjacent structures. There is a 9% to 14% risk of malignant recurrence in benign tumors, and long-term follow up care is important. Beyond the neonatal period, the risk of malignancy increases, and the tumor configuration is more likely to have an internal component (Altman types III and IV; see Fig. 63-13 ). Children with a large sacrococcygeal tumor suspected to be malignant should be evaluated for feasibility of resection. Gastrointestinal and genitourinary complications are far more common than lower extremity neurological sequelae, with up to 41% patients affected in one study. Long-term follow up observation is mandatory in these children to ensure that normal bowel, bladder, and sensory-motor function is preserved. If a problem is encountered, these children are best cared for by a multidisciplinary team that simultaneously initiates symptom management (such as management of constipation) with investigational studies including functional evaluations such as anorectal manometry and urodynamic studies (including direct electromyography [EMG] evaluation of the urinary sphincters) to define the extent of the problem at discovery as well as any progress (or lack thereof) in response to the prescribed interventions. One final note is recognition of the recommendation by Ozkan and colleagues, who suggest the need for defining the extent of neurologic, gastrointestinal, and genitourinary dysfunction before any intervention so as to better categorize any impairment of function at baseline and not just after surgery. This suggestion is especially valuable for those patients whose tumors are discovered outside of the neonatal period.
Therefore an initial biopsy with a delayed resection must be seriously considered if morbidity is likely to be reduced. In patients whose lesions involve the rectum or extend into the sacral bone or in patients already shown to have metastatic disease, biopsy rather than resection is the more appropriate initial surgical approach. If a needle biopsy is used to obtain diagnostic material, multiple passes should be obtained because of the histologic heterogeneity of these tumors and the possibility that the malignant foci will not be found in every sample. The histologic diagnosis should also be evaluated in the context of the information from tumor markers. A benign histologic diagnosis in the setting of elevated tumor markers should lead one to question whether there was a sampling error in the sample obtained or in the pathologic review. Lin28 is a new histochemical reagent that can be used to detect YST in the specific and sensitive way that should be used to detect occult YST.
Delayed resection of a SCT has been definitively demonstrated not to adversely affect survival. In fact, German investigators have shown that patients with locally advanced and metastatic tumors actually had better overall survival when treated with chemotherapy before tumor resection. Overall survival for patients with delayed resection was 83%, whereas overall survival for patients who had an initial resection was only 45% (P = 0.01). Preoperative chemotherapy allowed for more complete primary resection of the tumor; 19 of 31 patients had a complete resection after preoperative chemotherapy vs. only 11 of 35 patients who had a complete resection when definitive surgery was attempted before chemotherapy. Complete resection of the tumor, either at diagnosis or delayed until after chemotherapy, was the most significant predictor of event-free survival (EFS) in the German series of sacrococcygeal tumors ( Fig. 63-14 ). Second-look surgery was recommended for those with incomplete resection and had therapeutic benefit. Analysis of the data from the United States Pediatric Intergroup Trial (POG9049/CCG8882) also confirms that delayed resection did not have an adverse effect on prognosis.
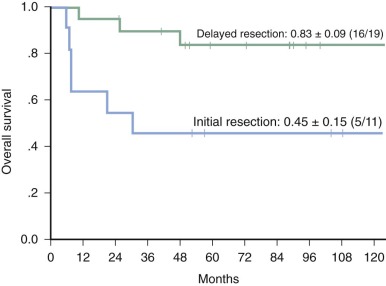
Most presacral lesions will be initially approached through a posterior transsacral incision. Superior extension of the tumor into the pelvis often requires a laparotomy. If laparotomy is performed, retroperitoneal lymph nodes should be examined and biopsied if enlarged. In all cases, the coccyx must be completely removed en bloc with the primary tumor. The importance of the resection of the coccyx was first noted by Gross, who reported a recurrence rate of 38% of those who had not had coccygectomy and 0% of those who had had a coccygectomy.
Surgical Approach to Testes Tumors
Risk of malignancy is age dependent with 38% to 74% of testicular tumors in the prepubertal age group having benign histology. In prepubertal boys with localized tumors, those with normal tumor markers and imaging studies can be considered for testis sparing enucleation of the tumor. Because the most common sites of metastasis of a testicular GCT are the retroperitoneal lymph nodes, a pelvic/abdominal CT scan should be obtained preoperatively. If the CT scan is delayed until after surgical exploration of the inguinal region, reactive retroperitoneal lymphadenopathy may be noted on CT scans and may be difficult to differentiate from metastatic disease. Evaluating the lungs for pulmonary metastases with chest CT before surgery is advisable, because postoperative atelectasis can make evaluation for malignant disease more difficult. Tumor markers should also be obtained preoperatively, because these levels are important to confirm the initial diagnosis and to provide prognostic information. In addition, the rate of decline is important to be able to track postoperatively in patients, especially in patients with stage I disease who are being expectantly observed, reserving chemotherapy only for those patients who have demonstration of residual malignant disease.
The surgical approach for a scrotal tumor should be through an inguinal incision. Control of the testicular vessels and vas deferens is gained at the level of the inguinal ring. The testis is then mobilized from the scrotum. Occasionally, a large testicular mass cannot be mobilized out of the scrotum into the inguinal region, and in this situation the inguinal incision should be enlarged to the superior aspect of the scrotum in order to avoid tumor rupture during mobilization. A radical orchiectomy is then performed with high ligation of all cord structures at the level of the internal inguinal ring. In prepubertal boys who have a focal lesion with normal markers and metastatic imaging, tumor enucleation protecting the tumor capsule followed by frozen section should be considered. If the frozen section is consistent with teratoma, the testis can then be placed back in the scrotal position, because enucleation is adequate therapy for prepubertal testicular teratoma. In the postpubertal boy, however, even localized teratoma has malignant potential and orchiectomy ought to be undertaken.
Rate of adherence to surgical guidelines was evaluated among the 63 patients with stage I GCT of the testes who were treated with observation only on the last U.S. intergroup trial from 1990 through 1996. Adherence to guidelines could be verified in only 69% of the patients. Likelihood of adherence was directly related to whether or not the surgeon thought the diagnosis was tumor preoperatively. In patients in whom the preoperative diagnosis was malignant tumor, the surgical guidelines were followed in 80% of patients, whereas among patients in whom another preoperative diagnosis was suspected (i.e., hydrocele, hernia, torsion), the guidelines were followed in only 17%. There was no difference in recurrence rate among those patients in whom the surgical guidelines were followed and among those for whom the guidelines were not followed, with the exception of patients who had had scrotal violations. Among the four patients with transscrotal violation, three had recurrence of disease.
Based on detailed review of the surgical procedures followed in the CCG-POG Intergroup Study (INT0098), the surgical guidelines in the case of scrotal violation were revised. In cases in which a scrotal orchiectomy has been performed without violation of the tumor capsule, the patient may still remain in stage I. If the cord structures show evidence of tumor spread, the patient should be upstaged to stage II. However, if a scrotal biopsy was performed as the initial procedure, the scrotum is considered to be contaminated and the patient should be treated as having stage II disease. Hemiscrotectomy, however, is no longer required. In addition, a “completion orchiectomy” with removal of all cord structures to the level of the internal ring should be performed.
Evaluation of Retroperitoneal Nodes in Pediatric Testicular Germ Cell Tumors
Lymphatic drainage of the testes is to the retroperitoneal nodes and to the nodes below the renal vessels. Right testicular tumors usually metastasize to the nodes between the aorta and the inferior vena cava (interaortocaval nodes). Left testicular tumors metastasize to the nodes lateral to the aorta (paraaortic). Left supraclavicular nodes and pulmonary nodules have been observed in the absence of retroperitoneal adenopathy. In adults, it is estimated that up to 30% of patients have false negative CT scans; in the most recent U.S. intergroup study of stage I testicular cancer among boys younger than 10 years of age, 5% of patients had failure of normalization of tumor markers postoperatively (i.e., patients had occult disease that was not detected on initial CT scan (false-negative initial CT scan). In total, 22% of patients presumed to be in stage I at diagnosis eventually relapsed, but all were salvaged with chemotherapy with 100% survival.
If the preoperative CT scan of the abdomen has identified enlarged (≥4 cm) retroperitoneal lymph nodes, biopsy is not necessary, and one may assume that this represents metastatic disease; the patient should be treated as having stage III disease. If retroperitoneal lymph nodes are between 2 and 4 cm, a biopsy should be performed. If the retroperitoneal nodes are smaller than 2 cm, a biopsy is not required; however, the tumor markers (AFP and β-HCG) should be observed and return to normal with appropriate decline according the half-life (see previous discussion of tumor markers). In adult males, 10% to 20% of nodes between the size of 1 and 2 cm contain metastatic disease.
If the decision is made to biopsy a suspected node of the prepubertal boy, the retroperitoneal procedure should simply be a lymph node excision, not a full retroperitoneal lymph node dissection. If no biopsy is performed and the nodes are enlarged (>2 cm), that patient should be treated as a patient with stage III disease. But in adolescents (as in adults), if a biopsy is being undertaken to evaluate nodal disease, then a nerve-sparing modified template retroperitoneal lymph node dissection (RPLND) should be employed, which some surgeons would expand to a full bilateral nerve-sparing RPLND if any of the initial nodes are positive on frozen section. RPLND is both diagnostic and potentially therapeutic. For further discussion of RPLND please see the section on treatment of adolescent boys with stage I testicular cancer.
Metachronous Testicular Cancer
In adults, 3% to 5% of men with testicular cancer have a metachronous tumor in the other testicle at diagnosis, and 2% of men subsequently develop a tumor in the contralateral testicle. These risks have not been quantified for pediatric patients and no formal evaluation of the contralateral testis is recommended besides that continued thorough physical examinations, although a patient should be advised of the likely need for continued surveillance of the contralateral testis. Patients who have developed a testicular tumor in a cryptorchid testis are at increased risk of malignancy in the normally descended contralateral testes.
Surgical Approach to Ovarian Tumors
The largest series of ovarian GCTs (66 cases) estimated rate of malignancy in an ovarian mass as 19% at ages 0 to 5 years, 14% at ages 6 to 10 years, and 33% at ages 11 to 15 years. AFP and β-HCG should be determined preoperatively for ovarian tumors. If not done preoperatively, these markers should be sent from the operating room once the diagnosis is apparent. The goals of the initial exploration for a suspected ovarian tumor are to completely resect the tumor if it can be accomplished without sacrifice of adjacent organs and to completely evaluate the extent of disease.
Surgical guidelines for ovarian GCTs were originally based on the experience with ovarian epithelial carcinoma, in which aggressive surgical resection is associated with higher chance of survival. Application of those surgical guidelines to ovarian GCTs, which have a different pattern of spread and are also highly chemosensitive, is not warranted. It is clear from studies in adult women with GCTs that fertility-sparing surgery (i.e., performing an unilateral instead of bilateral salpingo-oophorectomy) had no adverse outcome on survival. On the most recently completed U.S. Intergroup Trial for gonadal tumors (CCG8892/POG9048), surgeons adhered to the guidelines (derived from adult epithelial cancer) in only 2% of the patients, but overall EFS and overall survival (OS) rates were nonetheless excellent. Surgical omissions resulting in protocol noncompliance resulted from failure to biopsy bilateral nodes (97%), no omentectomy (36%), no peritoneal cytology (21%), and no contralateral ovary biopsy (59%). Among the patients in whom peritoneal cytology was examined, 25% of patients had malignant tumor found in what would otherwise been declared to be stage I disease. Biopsy of normal-appearing lymph nodes, omentum, and/or contralateral ovary very rarely, if ever, showed occult malignant disease.
Given the excellent overall survival, despite low adherence to the surgical guidelines on the Pediatric Intergroup Trial, the current surgical guidelines for pediatric ovarian GCT have been substantially revised. Given the high rate of positivity in the examination of peritoneal fluid, all patients with ascites present should have the fluid collected for cytology. In the absence of ascites, peritoneal washings should be conducted and sent for pathologic review. Any patient in whom cytology is not available should not be considered to be in stage I. The pelvic viscera should be examined, and pelvic and retroperitoneal lymph nodes should be palpated bilaterally. Only suspicious or enlarged lymph nodes should be biopsied. The omentum and peritoneal surfaces should also be palpated and visualized. If the omentum is adherent or has nodules or implants, partial or complete omentectomy that includes the lesions should be done. If the omentum does not appear grossly involved, an omentectomy does not need to be performed. Any nodules studding the peritoneal surfaces should be biopsied.
If only one ovary is involved, the tumor should be removed by unilateral oophorectomy. Attempts to preserve completely uninvolved fallopian tubes and the uterus should be made in all patients. Lesions adherent to the uterus may be managed by separation of the adherence if feasible or by biopsy only with planned second-look surgery after chemotherapy if necessary. The tumor capsule should not be aspirated. If the capsule is violated, either by the surgeon or because of rupture by the tumor or if there is microscopic capsule penetration seen by the pathologist in an apparently intact tumor, the patient should be upgraded to stage II (this is in contrast to adult staging guidelines in which a patient with capsular involvement is International Federation of Gynecology and Obstetrics [FIGO] stage IC). The ovary must be delivered intact and not removed in fragments. The contralateral ovary should only be biopsied if it appears involved with tumor; biopsy of a normal-appearing contralateral ovary is discouraged because of concern regarding possible adverse impact on future fertility. Bilateral ovarian lesions were present in only 6% of patients (n = 10) on the U.S. Pediatric Intergroup Study. Of these, four were benign teratomas and six were malignancies. If a patient has bilateral disease, preservation of as much normal ovarian parenchyma as possible should be the goal.
Surgical Approach to Mediastinal Germ Cell Tumors
The German MAKEI experience with mediastinal GCTs was analogous to the experience with sacrococcygeal tumors. Delayed resection increased the odds of complete resection of the primary tumor (10 of 11 patients at delayed resection vs. 6 out of 12 patients at primary resection) and complete resection was a strong predictor of more favorable outcome (EFS 0.94 ± 0.06 vs. 0.42 ± 0.33). Therefore the surgeon must decide whether it is more prudent to attempt a primary resection or to obtain a biopsy and plan for delayed resection.
The resection may be through a lateral thoracotomy or median sternotomy, as determined by the site of the lesion. The margin of resection should include adherent nonvital structures such as the thymus and pericardium. Regional lymph nodes should be evaluated and biopsied when possible in all patients.
Surgical Approach to Extragonadal Tumors at Other Sites
YST (also referred to as endodermal sinus tumor ) of the vagina is a rare tumor occurring exclusively in children younger than 3 years of age. The mass can be confused with sarcoma botryoides. Only small lesions that can be resected with vaginal preservation should be excised at initial presentation. In others, a careful vaginal examination and limited biopsy should be performed. After completion of chemotherapy, repeat evaluation and complete excision of residual disease should be considered, although oftentimes residual tissue is just scar tissue. A biopsy to guide the need for resection is a reasonable approach.
Malignant GCTs found at other locations should be completely excised if possible without sacrifice of adjacent organs. Biopsy only may be appropriate at the initial operation, with planned secondary procedures for delayed excision after chemotherapy. Any enlarged regional lymph nodes in the area should be sampled.
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
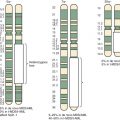 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


