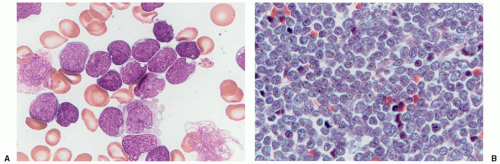
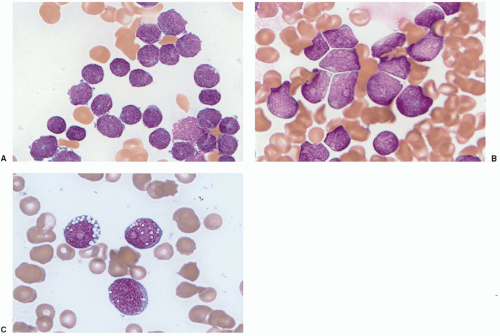
Acute lymphoblastic leukemia is the most common hematopoietic tumor of childhood, and is defined as a proliferation of immature lymphoid cells or lymphoblasts. A minimal threshold for defining leukemia has not been established by the WHO for ALL, in contrast to the 20% blasts used for AML. Protocols may require lymphoblasts to comprise 25% or more of bone marrow cells. The diagnosis is rarely made in the setting of less than 20% blasts. ALL is biologically similar to lymphoblastic lymphoma, distinguished by evidence of extramedullary disease and less than 25% bone marrow lymphoblasts in B-lymphoblastic lymphoma. The most clinically and biologically relevant classification schemes for ALL incorporate molecular genetic or cytogenetic features, in conjunction with immunophenotype. Lymphoblast morphology is not helpful in subclassifying ALL into clinically important prognostic groups. Burkitt leukemia/lymphoma, the older French-American-British (FAB) L3 type, is classified as a mature B-cell non-Hodgkin lymphoma and should not be termed B-ALL.
Detection of B-cell immunoglobulin or T-cell receptor gene rearrangements is not lineage-specific in the precursor lymphoid neoplasms as both types of rearrangements may occur in the same cell type. B-ALL characteristically expresses the B-cell markers CD19 and CD79a, as well as the immature lymphoid marker TdT. Most cases are CD10 positive, but there is variation in the expression of CD45, CD20, or CD34. Because these are neoplasms of immature B-lineage cells, they do not usually express surface immunoglobulin heavy or light chains; however, the presence of restricted immunoglobulin light or heavy chains in the presence of an otherwise immature cellular immunophenotype (TdT-positive) does not preclude a diagnosis of precursor B-cell ALL. Immunophenotyping discriminates B- and T lymphoblasts from minimally differentiated AML. Immunophenotyping is also useful for distinguishing residual or recurrent B-ALL from the presence of hematogones (benign precursor B cells) in the bone marrow. Minimal
residual disease monitoring may use multidimensional flow cytometry, patient-specific molecular assays for the immunoglobulin gene rearrangement of the leukemic clone, or the presence of a characteristic gene fusion or mutation.
The majority of B-lymphoblastic neoplasms present as leukemia, fewer cases present with extramedullary disease, and less than 25% present as blasts in the bone marrow (B-lymphoblastic lymphoma). The recurrent genetic abnormalities common to most cases of B-ALL are less often associated with B-lymphoblastic lymphoma. These cases have not been studied in molecular detail, but it appears that additional copies of regions of chromosome 21, particularly the RUNX1 region at 21q22, may be pathogenic.
B-Lymphoblastic Leukemia
The 2008 WHO recognizes seven recurrent genetic aberrations (
Table 7.2).
5 The incidence of each subtype varies by age. These recurrent genetic factors are an important component of risk stratification, together with factors such as patient age, presenting white blood cell (WBC) count, and measures of early treatment response.
6 Array-based molecular karyotyping identifies multiple submicroscopic alterations in many of these groups that may further influence disease risk and treatment in the future.
7The Philadelphia chromosome resulting from the (9;22) (q34;q11.2) chromosomal translocation is found in chronic myelogenous leukemia (CML), adult ALL, and 4% of pediatric B-ALL. The BCR/ABL1 fusion protein of pediatric ALL (p190) is smaller than that of CML (p210). Five to ten percent of cases display normal karyotypes, with a
BCR/ABL1 fusion detectable by molecular methods. ALLs with t(9;22) are of precursor B-cell lineage (CD10, CD19, and TdT positive) and may show aberrant expression of the myeloid-associated antigens CD13 and CD33, as well as CD38. The 2008 WHO classification proposes strict criteria to distinguish B-ALL with t(9;22) from mixed phenotype acute leukemia (MPAL) with t(9;22), mainly based on evidence of myeloperoxidase (MPO) in the blasts of MPAL.
8 Deletions of
IKZF1, resulting in oncogenic Ikaros isoforms, are common in
BCR-ABL1-positive B-ALL.
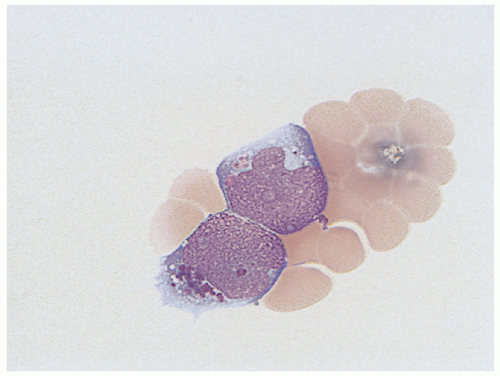
9B-lymphoblastic leukemia with
MLL (now known as
KMT2A) abnormalities, particularly t(4;11)(q21;q23), is the most common ALL type in infants, and constitutes approximately 5% of ALLs in older children or adults (
Fig. 7.4).
10 The blast immunophenotype is typically CD10 negative with aberrant expression of myeloid-associated antigens CD15 or CD65. Other myeloid antigens are not usually expressed (specifically, no more than one marker of monocytic differentiation may be present, such as NSE, CD11c, CD14, CD64, or lysozyme), and the blast cells are routinely MPO negative. These features are necessary to distinguish B-ALL from the diagnosis of MPAL with t(v;11q23)—
MLL rearranged. High-level expression of the FLT3 receptor tyrosine kinase within the leukemic blasts is a feature of this genetic subtype of ALL; however,
FLT3-activating mutations are found in only a small subset of cases.
11B-ALL with t(12;21) (p13;q22)
ETV6-RUNX1 is the most common genetic subtype of ALL in children, particularly in the 2- to 10-year age group, and constitutes at least 20% to 30% of childhood ALL.
5 The t(12;21) fuses the
ETV6 gene (aka
TEL) on chromosome 12 with the
RUNX1 (aka
AML1) gene on chromosome 21. This aberration is not detectable by routine karyotype analysis and requires either RT-PCR or FISH evaluation for detection. The leukemic blasts are of precursor B-cell lineage (CD10, CD19, and TdT positive), lack expression of CD9 and CD20, and may show aberrant expression of CD13.
Blast clones with an abnormal hyperdiploid karyotype of more than 50 chromosomes (high hyperdiploid) are seen in 25% of childhood ALL cases. The identification of this subgroup can be made either by routine cytogenetics or by assessing DNA ploidy using flow cytometry. Nearly all hyperdiploid ALLs have trisomy of chromosomes 6, tetrasomy of chromosome 21, and duplication of an X chromosome. Other chromosomes that are frequently trisomic include 4, 10, 14, 17, and 18.
12 No specific immunophenotypic
features are recognized. FLT3-activating mutations have been identified in 10% to 20% of cases.
11Hypodiploid ALL is defined differently by different authors. The WHO assigns this diagnosis in B-ALL when the chromosome number is less than 46.
5 However, stricter definitions of less than 44 chromosomes may be more clinically significant.
13,14 Leukemias with less than 44 chromosomes are a rare subset comprising less than 1% of B-ALL, with a particularly poor prognosis. Near-haploid (23 to 29 chromosomes) or low-hypodiploid B-ALL can be missed by standard karyotyping if the hypodiploid clone has undergone endoreduplication, doubling the number of chromosomes. This discrepancy can be resolved by FISH. Molecular genetic studies of hypodiploid ALL identify unique differences from other ALL subtypes. Most near-haploid cases harbor activating mutations of Ras signaling (
NFI, NRAS, KRAS, and
PTPN11) and inactivating deletions and mutations of the
IKZF3 (AIOLOS).
Nearly all low-hypodiploid cases have mutations of
TP53 as well as inactivating mutations of
IZKF2 (HELIOS). The
TP53 mutations of low-hypodiploid ALL were also found in nontumor cells, suggesting possible Li-Fraumeni syndrome in many cases.
7B-ALL with t(5;14)(q31;q32)—IL3-IGH is a rare subtype of B-ALL occurring in both children and in adults.
5 Patients may present with ALL or asymptomatic eosinophilia. In the setting of eosinophilia, even small numbers of B lymphoblasts should prompt consideration of this entity and cytogenetic study to confirm the diagnosis. These cases should be distinguished from the rare lymphoblastic leukemias with mutations of
PDGFRA or
FGFR1 commonly associated with eosinophilia.
B-ALL with a balanced or unbalanced t(1;19)(q23;p13.3)
TCF3-PBX1 chromosomal translocation occurs in approximately 6% of pediatric ALL, with an average age of 5 years. A rare variant chromosomal translocation t(17;19) fuses TCF3 and HLF. Up to 25% of cases with
TCF3/PBX1 fusions detected by molecular genetic studies may have normal routine karyotypes.
7 The blasts are of precursor B-cell lineage (CD10, CD19, and TdT positive), express cytoplasmic mu and CD9, and usually lack CD20 and CD34.
B-ALL lacking one of the aforementioned recurrent genetic abnormalities is categorized as B-lymphoblastic leukemia/lymphoma, not otherwise specified (NOS) by the 2008 WHO classification.
15 Additional subtypes have emerged from molecular genetic studies. Some have prognostic significance that may be useful in risk-based stratification of therapy. These include the following: (1) B-ALL with intrachromosomal amplification of chromosome 21 (iAMP21), characterized by gain of at least three copies of a region of chromosome 21 that always includes
RUNX1; this may be seen most frequently in B-ALL, NOS, but also occurs in cases with
ETV6-RUNX1 and
BCR-ABL1 rearranged; (2) ERG-altered B-ALL, occurring exclusively in B-ALL; NOS; (3)
CRLF2 rearranged B-ALL, with rearrangement of the cytokine receptor gene
CRLF2 at Xp22.3/Yp11.3 occurring in 50% of Down syndrome (DS)-associated ALL; (4)
BCR-ABL1 like B-ALL, characterized by a similar gene expression profile as B-ALL with
BCR-ABL1 but lacking the translocation, with numerous associated genomic alterations.
7
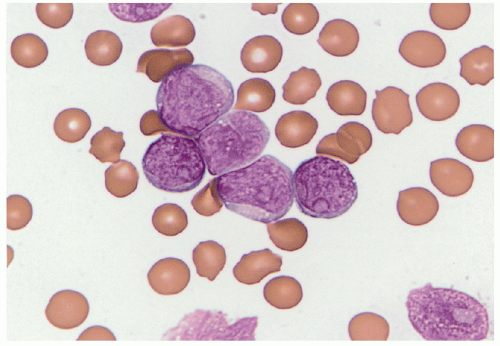
T-Lymphoblastic Leukemia/Lymphoma
T-lymphoblastic tumors constitute approximately 15% of pediatric lymphoblastic leukemia and 85% to 90% of cases that present as lymphoblastic lymphoma.
16 Cases with 25% or more bone marrow involvement are considered to be ALL. Approximately half of T-ALL patients have a mediastinal mass at the time of presentation, and other organ sites may be involved. Immunophenotyping is required to differentiate precursor T lymphoblasts from precursor B lymphoblasts. T-lymphoblastic neoplasms are TdT positive and usually express CD2, cytoplasmic CD3, CD5, and CD7, although loss of one or more of these T-cell antigens is common. Some cases also express CD1a, CD4, CD8, dual CD4 and CD8, or surface CD3. Surface and/or cytoplasmic CD3 expression is considered the most specific marker for the T lineage. T lymphoblasts also occasionally express CD79a (a B-lineage marker) or aberrantly express myeloid antigens such as CD13, CD33, or CD117.
The 2008 WHO classification does not further classify T-ALL by recurring cytogenetic abnormalities. A total of 50% to 70% of T-ALL have an abnormal karyotype, most commonly involving the T-cell receptor loci. Detection of these aberrations at diagnosis may be useful for disease monitoring, but distinct prognostic disease groups defined by recurring cytogenetic abnormalities are not yet part of any major classification scheme for risk group stratification.
17 Mutations of
PHF6, NOTCH1, and
FBXW7 have been found in several cohorts of T-ALL, but a consistent role for these mutations in risk stratification has not been identified to date.
7 One subtype of T-ALL, early T-cell precursor (ETP) ALL has been characterized immunophenotypically by expression of cytoplasmic or surface CD3, dim CD5, absence of CD8 or CD1a, and coexpression of CD34, CD117, HLA-DR, CD13, or CD117. ETP-ALL appears to have a poor prognosis, the karyotype may be complex, and numerous diverse genetic alterations are found. The transcriptional profile resembles that of hematopoietic stem cells and primitive AML more than that of human precursor T cells.
7,18