Fig. 9.1
Linear representation of F8, the structure of FVIII, and its cleavage site. The 26 exons (above) and domain organization (below) of FVIII based on amino acid homology. Activation of FVIII leads to release of the B and a3 domains. In the activated FVIII heterotrimer, the A1 and A3 domains retain the metal ion-mediated interaction, and the stable A1/A3-C1-C2 dimer is weakly associated with the A2 subunit through electrostatic interactions. Either spontaneous dissociation of A2 or proteolysis of FVIIIa results in loss of its function. Th thrombin
Removal of a 19 peptide secretory leader sequence of FVIII results in the mature sequence of 2332 amino acids. The FVIII molecule consists of three homologous A domains, a B domain, and two homologous C domains structurally in the order A1-A2-B-A3-C1-C2. Prior to secretion, FVIII is processed into a series of metal ion-linked heterodimers produced by cleavage at the B-A3 junction together with a number of additional cleavages within the B domain. FVIII is highly sensitive to proteolytic processing after secretion, and only a small fraction of circulating FVIII retains a single-chain form. Most consist of heavy chains of variable length (including sequences of the A1, A2, and B domains) linked ion covalently to light chains composed of the A3, C1, and C2 domains [4]. Limited proteolysis at Arg372 and Arg1689 by thrombin or FXa generates cofactor activity (FVIIIa). Expression of recombinant (r)FVIII lacking the entire length of the B domain has confirmed, however, that this domain is dispensable for the activation and procoagulant function of the protein. The cofactor function of FVIIIa accelerates the activation of FX by activated FIX (FIXa) on a suitable phospholipid surface, thus amplifying the clotting stimulus by several orders of magnitude. Specific proteolytic cleavages between the FVIII domains both activate and inactivate the cofactor [5]. The active form (FVIIIa) cation-dependently bonds to the A3-C1-C2 light chain together with the heavy chain held by electrostatic association between the A1 and A2 domains. FVIIIa is highly unstable owing to spontaneous dissociation of the A2 subunit and proteolytic inactivation of FVIIIa through cleavage at Arg336 by activated protein C (APC) and FXa. An additional cleavage site for APC at Arg562 in the A2 subunit results in complete inactivation of FVIIIa [5].
For convenience, F8 defects associated with hemophilia A may be divided into several categories: (1) gross gene rearrangements, (2) deletions or insertions of genetic sequence of a size varying from one base pair up to the entire gene, and (3) single DNA base substitutions resulting in either amino acid replacement (“missense”), premature peptide chain termination (“nonsense” or stop mutations), or mRNA splicing defects. More than 2500 unique molecular defects in F8 have been described and are included in the worldwide Factor VIII Variant Database (http://www.factorviii-db.org). Molecular characterization of patients with hemophilia A is dependent initially upon the analysis of intron 22 and intron 1 in F8 [6, 7]. The single, clinically most important defect is a gene rearrangement (an inversion) involving intron 22 (Fig. 9.2), which is evident in approximately 50% of all severe disease cases [6]. If genetic variations in intron 1 or intron 22 are not detected, full F8 mutation screening is generally performed by direct sequencing, covering all exons, intron-exon boundaries, and the promoter region. Genetic variations have not been identified in 2–18% of patients [8]. Consistent gene defects in patients with mild and moderate FVIII deficiencies are not common, and full mutation analysis of F8 by direct sequencing is usually necessary in these patients.
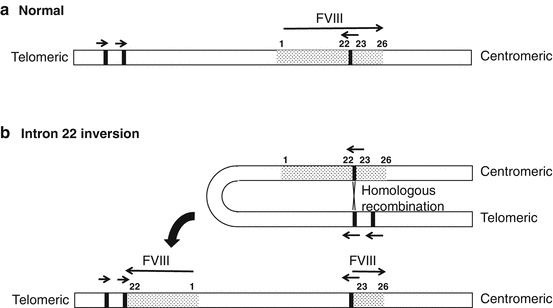
Fig. 9.2
Simplified representation of the intron 22 inversion. Recombination between homologous sequences in intron 22 and ~400 kb telomeric to the gene leads to separation of exons 1–22 from exons 23–26
9.1.3 FVIII Inhibitors
The presence of neutralizing alloantibodies (inhibitors) to therapeutic FVIII is a serious complication in hemophilia A, occurring in approximately 30% of patients classed as severe [9]. In those individuals, the inhibitors usually develop during the first 20–30 days of exposure and appear to result from a multi-causal immune response involving both patient-related and treatment-related factors. A gene defect is thought to contribute to about 40% of the risk of inhibitor formation, although the immunology of inhibitor development remains to be fully understood. Large deletions and nonsense mutations seem to present the highest risk, whereas missense and splicing mutations mediate the lowest risk [10]. The most common acquired risk factor is the intensity of exposure to FVIII concentrate [11]. In non-severe hemophilia A patients and different from severe hemophilia A, inhibitors usually arise when the immune system is under intense stimulation or when exposure to FVIII concentrates is unusually high, independent of exposure days, in particular in the postoperative period [12, 13]. Mutations leading to an abnormal conformation of FVIII are associated with a high risk of inhibitor development in mild hemophilia A [13], especially in those patients with mutations in the A2 and A3C1C2 domains (e.g., Arg593Cys and Arg2150His, respectively) [14, 15].
9.1.4 Treatment
Comprehensive guidelines for the treatment of hemophilia were endorsed as high standard by the International Society on Thrombosis and Hemostasis (ISTH) in 2012 (https://www.isth.org/?page=Published_Guidance) and were published in detail in 2013 by the World Federation of Hemophilia (WFH) working group [16]. The primary aim of hemophilia care is to prevent and treat hemorrhage using FVIII therapeutic products. An integrated support system, called comprehensive care, is highly recommended and includes specific therapy for prophylaxis or acute bleeding and also treatment for complications and drug-related side effects [17]. As noted above, however, the development of inhibitors presents many difficulties for the use of FVIII concentrates, and long-term complications, such as joint destruction with the need for synovectomy or joint replacement, together with infections transmitted by clotting factor concentrates are also important issues for maximally effective clinical management. Treatment for hemophilia A may not be straightforward, therefore, and depends overall on the severity of the FVIII defect, the presence of inhibitors, available resources, and clinical complications.
9.1.4.1 Replacement Therapy
Acute bleeding should be treated immediately with FVIII concentrate. Home infusion therapy has made it possible for patients to treat themselves quickly just after bleeding (and prophylactically). In the absence of an inhibitor, each unit of intravenously infused FVIII per kilogram of body weight will raise the plasma FVIII level by approximately 2 IU/dL [17]. The half-life of FVIII is 8–12 h, and optimal doses and duration of treatment are dependent on the site of bleeding and individual patient pharmacokinetics (Table 9.1) [16].
Table 9.1
Summary of the desired plasma factor peak levels and duration of administration (modification of [16])
Type of hemorrhage | Hemophilia A | Hemophilia B | ||
|---|---|---|---|---|
Desired level (IU/dL) | Duration (days) | Desired level (IU/dL) | Duration (days) | |
Joint | 40–60 | 1–2 | 40–60 | 1–2 |
Superficial muscle | 40–60 | 2–3 | 40–60 | 2–3 |
Iliopsoas and deep muscle | ||||
Initial | 80–100 | 1–2 | 60–80 | 1–2 |
Maintain | 30–60 | 3–5 | 30–60 | 3–5 |
CNS/head | ||||
Initial | 80–100 | 1–7 | 60–80 | 1–7 |
Maintain | 50 | 8–21 | 30 | 8–21 |
Throat and neck | ||||
Initial | 80–100 | 1–7 | 60–80 | 1–7 |
Maintain | 50 | 7–14 | 30 | 7–14 |
Gastrointestinal | ||||
Initial | 80–100 | 7–14 | 60–80 | 7–14 |
Maintain | 50 | 30 | ||
Renal | 50 | 3–5 | 40 | 3–5 |
Deep laceration | 50 | 5–7 | 40 | 5–7 |
Surgery (major) | ||||
Pre-op | 80–100 | 60–80 | ||
Post-op | 60–80 (1–3 day), 40–60 (4–6 days), 30–50 (7–14 days) | 40–60 (1–3 day), 30–50 (4–6 days), 20–40 (7–14 days) | ||
Surgery (minor) | ||||
Pre-op | 50–80 | 50–80 | ||
Post-op | 30–80 | 1–5, depend on the procedure | 30–80 | 1–5, depend on the procedure |
Hemarthrosis is a common complication in hemophilia patients, and repeated hemorrhage often occurs into the same joint, commonly called a target joint. In addition, compartment syndrome is a painful and potentially serious condition caused by bleeding or swelling within an enclosed bundle of muscles, and this poses a particular risk in hemophilia when soft tissue hemorrhage occurs [18]. The WFH strongly recommends the use of viral-inactivated plasma-derived (pd) or recombinant (r)FVIII concentrates in preference to cryoprecipitate or fresh frozen plasma for the treatment of hemophilia A [16]. rFVIII products especially have the theoretical advantage of being free from blood-borne pathogens. Recent research has also focused on inhibitor risk in patients treated with pd-FVIII or rFVIII, but the outcomes remain controversial. The Research of Determinants of Inhibitor Development (RODIN) study group has comprehensively examined the association between FVIII products and inhibitor development, and the only significant result determined was an unexpectedly higher risk of inhibitor development using second-generation full-length recombinant products compared with third-generation rFVIII products [19]. A more recent report [20] indicated that the risk of developing an inhibitor when using rFVIII products in previously untreated patients (PUPs) was significantly higher than when using pd-FVIII concentrates that contain von Willebrand factor (VWF), although this finding was not totally consistent with the earlier RODIN findings. Further research on inhibitor development is warranted to elucidate the precise immunogenicity of rFVIII.
Several clinical trials have been undertaken to investigate the use of rFVIII bioengineered to extended half-life (EHL), including Fc-fusion, PEGylated, and glycoPEGylated proteins. Fc-fusion proteins are designed to be recycled into the circulation through pH-dependent binding to neonatal Fc receptors (FcRn) that delay lysosomal degradation [21], and one such preparation, rFVIIIFc, is currently available [22]. Moreover, other EHL-modified rFVIII products, including BAX 855 (PEGylation) [23] and N8-GP (glycoPEGylation) [24], have also completed Phase 3 trials. All of these novel products appeared to face to a “ceiling effect,” however, and half-lives appear to be limited to approximately 1.5 to 1.6-fold of unmodified FVIII. This limitation could be related to the absence of VWF, the career protein.
For mild or moderately severe FVIII deficient patients, desmopressin (DDAVP) is an alternative option that rapidly increases FVIII:C by three- to sixfold from baseline levels [25]. DDAVP is generally administered intravenously at a dose of 0.3 μg/kg over 20–30 min and mediates elevated FVIII in 30–60 min that persists for 6–12 h. Tachyphylaxis occurs after repeated administration, however, due to depletion of endothelial stores [26].
9.1.4.2 Prophylaxis
Strategies for prophylaxis replacement therapy are based on evidence that moderately severe patients who have FVIII levels more than 1 IU/dL experience many fewer spontaneous bleeding events and have much better long-term joint function than those patients classed as severe [27]. In addition, randomized control studies have also convincingly demonstrated that prophylaxis with rFVIII can prevent joint damage and decrease the frequency of joint and other hemorrhages in young boys with severe hemophilia A [28]. Furthermore, even using prophylactic regimens that resulted in trough FVIII levels <1 IU/dL, episodes of joint hemorrhage were significantly decreased [29]. A range of different prophylactic protocols have been proposed, including 10–20 IU/kg or 25–40 IU/kg per dose 2–3 times a week, once a week for very young children [16], or temporal prophylaxis just before physical activity associated with a higher risk of injury [30]. It is clear, however, that the pharmacokinetics of FVIII is diverse, and the lowest effective level of FVIII should be determined individually for each patient considered for primary or secondary prophylaxis.
9.1.4.3 Adjunctive Therapy
Replacement therapy is the primary hemostatic treatment in patients with hemophilia. In addition, the concept of “RICE” (rest, ice, compression, and elevation) plays an important role as a first aid for acute bleeding in muscles and joints. Antifibrinolytics that enhance clot stability, such as tranexamic acid and epsilon aminocaproic acid, are also useful for mucosal and oral bleeding including dental extraction [31]. Tranexamic acid is commonly used at 25 mg/kg per dose every 6–8 h intravenously or orally. Physical fitness and the development of strong muscles are also recommended to protect joints. An appropriate exercise lifestyle should be encouraged, especially for children, to avoid overweight or obesity [32]. Noncontact sports such as swimming should be encouraged. In the absence of thoroughly controlled prophylaxis, high-contact sports including football or high-velocity activities including skiing should be avoided due to the obvious potential for life-threatening injuries.
9.1.4.4 Inhibitors
Hemostatic Management
Anti-FVIII alloantibodies occur about 30% of patients with hemophilia A and remain a challenging clinical problem in congenital FVIII deficiency [9]. Acute bleeding in patients with inhibitors may be treated effectively with infusions of concentrates which “bypass” the intrinsic coagulation pathway. Two such bypassing agents are available, activated prothrombin complex concentrate (aPCC) and recombinant FVIIa (rFVIIa), and both are safe and efficient almost equally [33], despite the differences in their content, dosing, and mode of action. Preference often seems to be dependent on individual patient circumstances, and combined use of both drugs is sometimes required [34]. Prophylaxis with aPCC in both adult and pediatric patients with inhibitors has been reported to be effective in preventing joint bleeds and longer-term orthopedic damage [35]. This type of treatment could be an important alternative choice for inhibitor patients who have failed or are ineligible for ITI.
Related posts:
 Inherited Bone Marrow Failure Syndrome, TAM
Inherited Bone Marrow Failure Syndrome, TAM
 Childhood Aplastic Anemia
Childhood Aplastic Anemia
 Hemophagocytic Lymphohistiocytosis, Secondary
Hemophagocytic Lymphohistiocytosis, Secondary
 Primary Hemophagocytic Lymphohistiocytosis
Primary Hemophagocytic Lymphohistiocytosis
 Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
 Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


