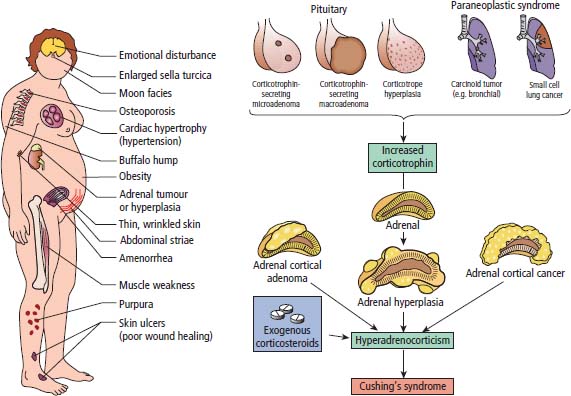
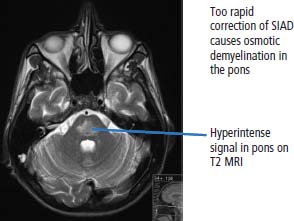
39 Paraneoplastic complications of malignancy are remote effects of cancer that occur without local spread. Most of these paraneoplastic syndromes arise due to secretion by tumours of hormones, cytokines and growth factors. Paraneoplastic syndromes also arise when normal cells secrete products in response to the presence of tumour cells. For example, antibodies produced in this fashion are responsible for many paraneoplastic neurological syndromes including cerebellar degeneration, Lambert–Eaton myasthenic syndrome and paraneoplastic retinopathy. Paraneoplastic neurological complications always appear on the list of differential diagnoses. However, just as viewers of House MD will know that “it’s not lupus”, it is rarely paraneoplastic either. Cushing’s syndrome is a clinical disorder resulting from prolonged exposure to excess glucocorticoids and should not be confused with Cushing’s disease which refers exclusively to those cases that arise due to an adrenocorticotrophic hormone (ACTH) secreting pituitary adenoma (Table 39.1). Clinically overt Cushing’s syndrome caused by ectopic secretion of ACTH by non-endocrine-derived tumours is rare. Approximately 20% of cases of Cushing’s syndrome are caused by ectopic ACTH secretion by a tumour that is frequently occult at presentation. For this reason the differential diagnosis between pituitary adenoma and ectopic ACTH is important clinically but biochemical overlap often makes this difficult. More than half the cases of ectopic ACTH syndrome are due to small cell lung cancer, with carcinoid tumours and neural crest tumours (phaeochromocytoma, neuroblastoma, medullary cell carcinoma of the thyroid) accounting for a further 15%. The typical presentation is of a middle-aged smoker with features of severe hypercortisolism and hypokalaemic metabolic alkalosis. Patients have muscle weakness or atrophy, oedema, hypertension, mental changes, glucose intolerance and weight loss. When ectopic ACTH production arises from a more benign tumour, for example, bronchial carcinoid or thymoma, the other classic features of Cushing’s syndrome may be present including truncal obesity, moon facies and cutaneous striae (Figure 39.1). The diagnosis of Cushing’s syndrome may be confirmed by elevated urinary free cortisol, loss of diurnal variation of plasma cortisol and failure of cortisol suppression in the low-dose dexamethasone (2 mg) test. After establishing the diagnosis, an elevated plasma ACTH supports the diagnosis of pituitary adenoma or ectopic ACTH syndrome. Failure of cortisol suppression following high-dose dexamethasone (2 mg four times daily for 2 days, or 8 mg overnight) and very high levels of ACTH (>200 pg/mL) suggest an ectopic source of ACTH. In difficult cases, a corticotrophin-releasing hormone (CRH) stimulation test, selective venous catheterization of the inferior petrosal sinus with ACTH estimations, somatostatin analogue scintigraphy and technetium-99 methoxyisobutylisonitrile (MIBI) imaging may be necessary to determine the source of ACTH. Table 39.1 Aetiology of Cushing’s syndrome ACTH, adrenocorticotrophic hormone; CRH, corticotrophin-releasing hormone. The mainstay of palliative therapy for Cushing’s syndrome due to ectopic ACTH production is inhibition of steroid synthesis, although inhibition of ACTH release and blocking glucocorticoid receptors have also been attempted. Several steroid synthesis inhibitors are available and successful use in these circumstances has been reported with aminoglutethamide, metyrapone, mitotane, ketoconazole and octreotide. On rare occasions laparoscopic bilateral adrenalectomy or adrenal artery embolization may be necessary to control symptoms. Hyponatraemia is a common finding in association with advanced malignancy and many factors may contribute including cardiac and hepatic failure, hyperglycaemia and diuretics. However, the detection of concentrated urine in conjunction with hypo-osmolar plasma suggests abnormal renal free water excretion and the presence of the syndrome of inappropriate antidiuresis (SIAD). This acronym is better than the previous term “syndrome of inappropriate antidiuretic hormone” (SIADH), since there is no vasopressin (ADH) secretion in approximately 15% of cases. In malignancy-related SIAD, tumours secrete ectopic arginine vasopressin or vasopressin-like peptides. In many respects SIAD is the opposite of diabetes insipidus; patients with SIAD are waterlogged with low plasma osmolality and sodium and high urine osmolality, whilst patients with diabetes insipidus are dehydrated with high plasma osmolality and sodium and low urine osmolality. SIAD is most frequently associated with small cell lung cancer or carcinoid tumours but has also been described in pancreatic, oesophageal, prostatic and haematological malignancies. Nonetheless many factors may contribute to SIAD (Table 39.2). Ecstasy (MDMA, methylenedioxymethamphetamine) is another cause of SIAD although over-drinking water at raves may contribute to the hyponatraemia too. Figure 39.1 Causes and clinical features of Cushing’s syndrome. Table 39.2 Causes of the syndrome of inappropriate diuresis (SIAD) ADH, antidiuretic hormone; SSRI, selective serotonin reuptake inhibitor. *Excessive water consumption may contribute to the development of hyponatraemia with ecstasy. Table 39.3 Diagnosis of the syndrome of inappropriate diuresis (SIAD) Significant symptoms of hyponatraemia appear at plasma sodium levels below 125 mmol/L, with confusion progressing to stupor, coma and seizures as levels fall. Nausea, vomiting and focal neurological deficits may also occur. The clinical features depend on both the levels of plasma sodium and the rate of decline. With gradual falls in sodium, the brain cells are able to compensate against cerebral oedema by secreting potassium and other intracellular solutes. Asymptomatic hyponatraemia therefore suggests chronic SIAD rather than acute SIAD. The division into chronic and acute SIAD is of therapeutic importance as their management differs. The diagnosis of SIAD requires the demonstration of plasma hyponatraemia and hypo-osmolality in the presence of concentrated urine and normal extracellular fluid volume (Table 39.3). The management of SIAD depends upon the rate of onset of hyponatraemia and the presence of neurological complications. Acute SIAD with an onset over 2–3 days and falls in serum sodium in excess of 0.5 mmol/L per day are associated with neurological sequelae and require prompt correction by intravenous hypertonic saline. However, correcting the plasma sodium too fast can also lead to severe neurological consequences including central pontine myelinolysis, an osmotic demyelination that causes acute paralysis, dsyphagia, dysarthria and coma (Figure 39.2). Hyponatraemia should not be corrected faster than 10 mmol/L/24 hours. In contrast, the mainstay of therapy for chronic asymptomatic SIAD is fluid restriction and inhibition of tubular reabsorption of water with drugs including the tetracycline antibiotic demeclocycline. Figure 39.2 Central pontine myelinolysis. Tumour-related hypoglycaemia is a frequent complication of β-islet cell tumours of the pancreas that secrete insulin (insulinomas), see Chapter 10, but occurs uncommonly with non-islet cell tumours. Most non-islet cell tumours produce hypoglycaemia through increased glucose use or by secreting insulin-like growth factors (IGF1 and IGF2). Non-islet cell tumours associated with hypoglycaemia are usually large retroperitoneal or intrathoracic sarcomas. Unlike other endocrine complications of malignancy, hypoglycaemia is very rarely associated with lung cancer. The clinical manifestations are due to cerebral hypoglycaemia and secondary secretion of catecholamines; they include agitation, stupor, coma and seizures that may follow exercise or fasting. Tumour-related hypoglycaemia should be differentiated from other causes of hypoglycaemia including drugs, for example, sulphonylureas, hypoadrenalism, hypopituitarism and liver failure. In advanced malignancy the most common cause of hypoglycaemia is continued oral hypoglycaemic medication in long-standing diabetics. Enteropancreatic hormone production is relatively uncommon in malignant disease. A variety of clinical syndromes occur associated with hormone secretion by endocrine tumours of the pancreas and less frequently tumours arising in other organs (see Chapter 10). The majority of pancreatic islet cell tumours are malignant with the exception of most insulinomas, and metastases are frequently present at diagnosis. For many patients the distressing clinical manifestations arising from excessive secretion of gastrointestinal peptides require palliation and this may be difficult to achieve. These tumours often secrete more than one polypeptide hormone and may switch their hormone production during follow-up. Carcinoid tumours arise from enterochromaffin cells principally in the gastrointestinal tract, pancreas and lungs but occasionally in the thymus and gonads (see Chapter 23). Chromaffin cells stain with or have an affinity for chromium salts, usually because the cells contain catecholamines. One in ten patients with carcinoid tumours develops the carcinoid syndrome after the development of hepatic metastases. This avoids the first pass metabolism of 5-hydroxytryptamine (serotonin, 5HT) and kinins in the liver so that the systemic symptoms occur. The acute symptoms are vasomotor flushing (typically of upper body lasting up to 30 minutes), fever, pruritic wheals, diarrhoea, asthma/wheezing, borborygmi and abdominal pain. Chronic complications include tricuspid regurgitation, arthropathy, pulmonary stenosis, mesenteric fibrosis, cirrhosis, pellagra (due to secondary deficiency of trytophan) and telangiectasia. The diagnostic investigation is 24-hour urinary collection of 5-hydroxyindole-acetic acid (5HIAA), a metabolite of 5HT. Somatostatin analogues are considered by most physicians to be the first-line treatment of choice for patients with carcinoid syndrome and indeed most enteropancreatic hormone syndromes. Palliation of the clinical manifestations of carcinoid syndrome includes symptomatic therapy of diarrhoea with codeine phosphate, loperamide or diphenoxylate, β2-adrenergic agonists for wheezing, and avoiding precipitating factors to reduce flushing such as including alcohol and some foods. Phaeochromocytomas arise from the chromaffin cells of the sympathetic nervous system, most frequently in the adrenal medulla but occasionally from sympathetic ganglia (see Chapter 22). Phaeochromocytomas commonly secrete norepinephrine (noradrenaline) and epinephrine (adrenaline) but in some cases significant quantities of dopamine are alsoproduced. Phaeochromocytomas are associated with a number of familial inherited cancer syndromes including multiple endocrine neoplasia (MEN) 2a, MEN 2b, von Hippel–Lindau syndrome and neurofibromatosis. The catecholamines cause intermittent, episodic or sustained hypertension and other clinical manifestations including anxiety, tremor, palpitations, sweating, flushing, headaches, gastrointestinal disturbances and polyuria. These symptoms are all attributable to excessive adrenergic stimulation.
Paraneoplastic complications of cancer
Paraneoplastic endocrine complications
Cushing’s syndrome
Type
Example
ACTH dependent
Pituitary adenoma (Cushing’s disease)
Ectopic ACTH secretion
Ectopic CRH secretion (very rare)
ACTH independent
Exogenous glucocorticoid administration
Adrenal adenoma
Adrenal carcinoma
Nodular adrenal hyperplasia
Syndrome of inappropriate antidiuresis
Source of ADH
Examples
Ectopic ADH production
Malignancy
Small cell lung cancer
Inappropriate pituitary secretion of ADH
Malignancy
Lung cancer Lymphoma
Inflammatory lung disease
Pneumonia Lung abscess
Neurological disease
Meningitis Head injury Subdural haematoma Surgery
Drugs
Antidepressants (tricyclics, SSRIs) Carbamazepine Chlorpropamide Phenothiazines Vincristine Cyclophosphamide Ecstasy*
Postoperative
Others
Hypothyroidism Porphyria Addison’s disease
Essential criteria to establish diagnosis
Plasma hypo-osmolality (plasma osmolality <275 mosmol/kg water and plasma sodium <135 mmol/L)
Concentrated urine (plasma osmolality >100 mosmol/kg water)
Normal plasma/extracellular fluid volume
High urinary sodium on a normal salt and water intake (urine sodium >20 mmol/L)
Exclude (i) hypothyroidism, (ii) hypoadrenalism and (iii) diuretics
Supportive criteria for diagnosis
Abnormal water load test (unable to excrete >90% of a 20 mL/kg water load in 4 hours and/or failure to dilute urine to osmolality <100 mosmol/kg water)
Elevated plasma arginine vasopressin levels
Non-islet cell tumour hypoglycaemia
Enteropancreatic hormone syndromes
Carcinoid syndrome
Phaeochromocytoma
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


