Historical background
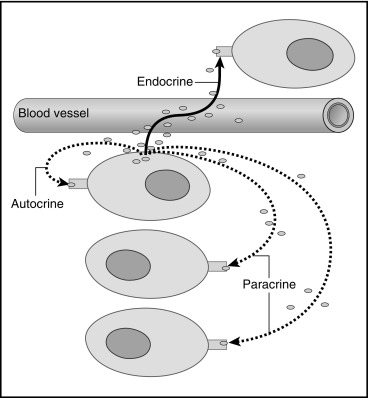
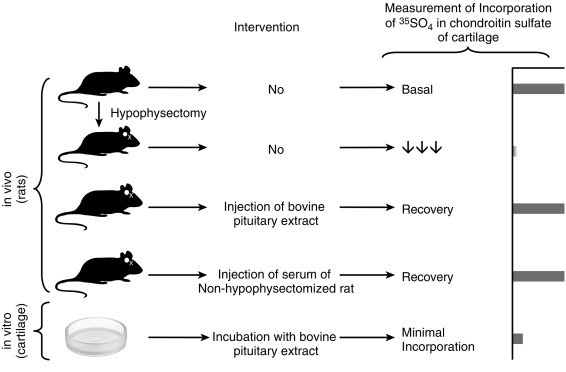
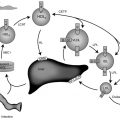
Endocrinology is a discipline of science that seeks to understand how chemical signals secreted by cells regulate the function of distant (endocrine) or local (paracrine) tissues, or even their own function (autocrine), to integrate vital processes of life, such as growth, reproduction, and metabolism ( Fig. 1.1 ). Classical endocrinology derived from careful clinical observation such as, for example, the gigantism associated with pituitary tumors, or the characteristic bodily changes now known as Cushing disease , which is also associated with pituitary tumors; histology indicated the former was likely the result of a product made by “acidophilic cells,” whereas the latter was associated with the expansion of “basophilic cells.” The products of these acidophilic or basophilic cells had to traverse the bloodstream to reach their distant and often multiple targets. Hence these were internal (“endocrine”) secretions, unlike the chemical substances secreted into ducts leading to a target tissue, for example, salivary glands into the cheeks; pancreatic enzymes into the duodenum (“exocrine”). Cushing disease was associated with hypertrophy of the adrenal cortex and certain tumors of adrenocortical tissues mimicked the features of Cushing disease. Therefore it was readily postulated that the pituitary secretes a substance that affects the adrenal glands and function; this substance was named adrenocorticotrophic hormone ( ACTH ) and it was deduced that the features of Cushing disease/syndrome were the result of a product or products from the adrenal gland. The destruction of adrenal tissue by tuberculosis or tumor was identified by Thomas Addison in 1855 and treatment of this entity with adrenal extracts, resulting in marked improvement, was first undertaken by William Osler in 1896. However, the purification of these “internal secretions” began in earnest only at the turn of the 20th century with spectacular success, as reviewed by Dr. Delbert Fisher. In the first quarter of the 20th century, epinephrine, thyroxin, insulin, and parathyroid hormone were purified, followed by the purification of the sex steroids from the ovary and testis, as well as the pituitary and placental gonadotropins that stimulated the gonads to secrete sex steroids. These purifications required laborious chemical methods and the elucidation or measurement of their function required costly and cumbersome bioassays. For example, the assay of the potency of insulin, discovered in 1921, required the use of rabbits; the definition of 1 international unit (IU) of insulin was assigned to be the amount of insulin that lowers the blood glucose of a healthy 2-kg rabbit, fasted for 24 hours, to 45 mg/dL or less, within 5 hours of injection. Clearly, such potency estimates reflected the relative crudeness of the purification; today’s pure recombinant human insulin possesses approximately 29 IU/mg, whereas the potency of porcine insulin in the early 1980s was around 23 IU/mg and likely less at the dawn of insulin therapy for diabetes mellitus. The bioassay of pure human insulin preparations has been abandoned; the purity of human insulin permits insulin to be now standardized as “one international unit of insulin (1 IU) is defined as the biological equivalent of 34.7 μg pure crystalline insulin.” It should be noted that 1 IU for insulin is not part of the International System of Units of the modern metric system, but is the pharmacologic International Unit as defined by the World Health Organization (WHO) Expert Committee on Biological Standardization. In addition to their cumbersome nature, the lack of sensitivity in the early bioassays prohibited the ability to measure insulin in normal blood or other biologic fluids; refinements, such as measuring the incorporation of labeled glucose into the fat pad or diaphragm of a rat, represented only an incremental improvement. Similarly, growth hormone (GH), isolated in 1944, was initially bioassayed by its ability to increase the width of the tibia growth plate in rats, after a defined period of injections, and by comparing the unknown relative to a dose response of known concentrations administered in vivo. Attempts to improve sensitivity and specificity led to the “sulfation factor-somatomedin hypothesis” ( Fig. 1.2 ), in which it was postulated that GH leads to the generation of a second substance, derived from the liver, which mediates the growth-promoting (somatotropic) effects and hence was named somatomedin . Subsequent studies demonstrated that this somatomedin substance was identical to a factor in serum which had insulin-like properties in vitro, and this insulin-like effect was retained even after all insulin was “quenched” by an excess of antibodies specific for insulin. The convergence of these two pathways eventually led to the discovery of the factor now known as insulin-like growth factor ( IGF )-1 and later as well as IGF-2 and a family of binding proteins that acted as carriers of the hormones in serum but also possessed biologic properties of their own, by which the actions of the IGFs were mediated and modulated. Despite these early bioassay limitations, the scientific curiosity of these chemical substances that regulated functions as diverse as blood pressure (epinephrine, cortisol), water metabolism (arginine vasopressin, cortisol), growth (GH, sex steroids, thyroid hormone), glucose (insulin, cortisol), and reproduction (sex steroids, follicle-stimulating hormone [FSH], luteinizing hormone [LH]), spurred the formation of medical societies focused on endocrine diseases. As detailed in the article by Fisher from which the following historical aspects are quoted, the Association for the Study of Internal Secretions was established in 1918 in the United States and renamed the Endocrine Society in 1952.
Pediatric endocrinology began as a subspecialty only in the 1940s with the establishment of endocrine clinics at the Massachusetts General Hospital and at Johns Hopkins. These programs attracted postdoctoral trainees who then established their own pediatric endocrine units in the burgeoning growth of academic medical centers in the 1950s and 1960s. In the United States, the Pediatric Endocrine Society, initially named the Lawson Wilkins Pediatric Endocrine Society ( LWPES ), was formed in 1972 and established as a subspecialty by the American Board of Pediatrics, with its first certification examination in 1978; there are more than 1000 board-certified pediatric endocrinologists today in the United States. The European Society for Pediatric Endocrinology was formed in 1966, followed by the Japanese Society for Pediatric Endocrinology in 1967 and the British Pediatric Endocrine Group in 1972, all preceding the LWPES in the United States. Several other regional pediatric endocrine groups were formed, including the Australian Pediatric Endocrine Group, the Sociedad Latino Americana de Endocrinologia Pediatrica, and the Asia Pacific Pediatric Endocrine Society. All of these groups now meet jointly every 4 years at an International Pediatric Endocrine Congress.
Impact of hormonal assays and molecular biology
Two discoveries revolutionized the field of endocrinology and led to an explosion of basic, as well as clinically relevant, therapeutic knowledge in the second half of the 20th century. The first was the development of radioimmunoassay by Yalow and Berson, reported for insulin in 1960. Here was a method for measuring the low concentrations of a hormone, using as little as 10 to 50 μL of a biological fluid in an accurate, reproducible way, with precision and sensitivity adequate for in vivo studies in humans or other species, as well as in vitro studies, such as the regulation of insulin secretion by nutrients, hormones, ions, and pharmaceutical agents in whole animals, including humans, in vivo, or in isolated perfused pancreas, or in isolated islets. This was followed by the rapid development of assays for various hormones and an explosion of discovery, including the distinction between absolute and relative insulin deficiency as the difference between “juvenile” and “maturity onset” diabetes now known respectively as Type 1 DM and Type 2 DM , the regulation of GH secretion in normal individuals at different ages and in clinical disorders of growth, the changes in thyroid function at birth and the possibility of screening for neonatal hypothyroidism, and the changes in gonadotropins and sex hormones during the process of normal and abnormal puberty and later in aging. The discovery and purification of the hypothalamic-releasing hormones for thyroid-stimulating hormone (TSH), FSH/LH, GH, and ACTH were made possible by these precise assays using (rat) pituitary cells, perfused by protein fractions derived from the hypothalami of animals. The discovery that a hormone produced in a cell could affect the function of its neighboring cell(s), without traveling through the bloodstream (paracrine action) or even its own function (autocrine), was also enabled by the use of these sensitive and precise tools, expanding our concepts of a hormone as a chemical messenger that influences, directs, and coordinates cellular functions throughout the body (see Fig. 1.1 ). Similar principles enabled the identification of the receptor molecules at the cell surface or in its cytoplasm that permit the hormone signal to be transduced to a message for turning biological processes on, or off, in specific tissues. Refinements using the principles of radioimmunoassay (RIA), but without radioactivity, as well as newer techniques of liquid chromatography coupled with mass spectrometry, are the bases of modern laboratory methodologies for hormone measurement, as well as for other chemical substances, such as drugs; examples of modern application of these methods, as well as the pitfalls in performance and interpretation, are reviewed in Chapter 4 . How these hormonal signals elicit a specific response after recognition and binding to a specific cell surface or cytoplasmic receptor, and the subsequent cascade of events known as the signal transduction pathways and their relevance to pediatric endocrinology are discussed in Chapter 3 . The notion that a hormone may not be capable of eliciting a response, despite high concentrations, was implicit in the entity labeled pseudohypoparathyroidism by Dr. Fuller Albright, but receptors and their signal transduction pathways were only systematically investigated beginning in the 1970s. These systematic studies, still ongoing, continue to identify the mechanistic pathways by which a hormone, after binding to its receptor, may elicit a response in one tissue but not in another. There may be other reasons why a hormone does not elicit an appropriate tissue response despite apparent high concentrations in the circulation. For example, an abnormal sequence in a hormone may prevent its full action at the receptor, and feedback control increases the hormone’s secretion, leading to high concentrations of partially functioning hormone with only minor or moderate impairment of function. Examples of such abnormalities include disorders of proinsulin conversion to insulin. 11 and OMIM #616214 Depending on the site of the abnormality in cleavage of proinsulin, both the measurement of insulin and proinsulin or its cleavage product C-peptide, may demonstrate higher than normal values that could be interpreted as “insulin resistance.” These studies also recognized that an activating mutation in a receptor will mimic the action(s) of a hormone, although the hormone concentration may be barely detectable, as exemplified by the precocious puberty in McCune-Albright syndrome or by an activating mutation of the LH receptor in males, which results in precocious puberty with high testosterone, but paradoxically very low concentrations of LH, an entity named testotoxicosis (see Chapter 18 ). In contrast, loss of function mutations cause the same clinical syndrome as hormone deficiency, even though the hormone concentration is markedly increased compared with normal, as exemplified in Laron syndrome, with poor growth and low IGF-1 concentrations, despite high circulating growth hormone concentrations (see Chapter 11 ). Thus the concentration of a hormone may not be directly related to its action. In summary, it is the ability to measure hormones, as well as intermediary molecules and metabolites, such as cyclic adenosine monophosphate at low concentrations (e.g., picomolar to nanomolar), in small volumes of biological fluids that enabled the rapid proliferation and understanding of endocrine regulation and function. It may be difficult for any reader who was not brought up in the era of bioassays to fully appreciate the impact of the application of the tool of RIA and its modifications, as various competitive protein binding assays without radioactive markers, to modern endocrine concepts and practice. The theoretical basis for the competitive binding assays, pitfalls in their interpretation, and their application to studies of receptor-ligand interactions are detailed in Chapter 4 .
The second revolution built on the discovery of the double helix by Crick, Watson, and Wilkins, a discovery for which they received the Nobel Prize in 1962. This discovery and its ongoing offspring propelled the ability to identify the molecular basis of cell function, the genes that regulate these processes, and the genetic mutations that underlie congenital or acquired disorders, including those of the endocrine system. Pediatric medicine is a particular beneficiary of these techniques because congenital malformations of the endocrine glands, the abnormalities of hormone signaling, as a result of defective hormone synthesis, hormone processing or receptor function to recognize and act on the hormonal signal are at the core of pediatric endocrinology, as reflected in this book. Chapters 2 and 3 provide an overview of the molecular and genetic methodologies applied in practice and research and also discusses the pitfalls in interpreting the results of genetic mutational analyses. This is a rapidly evolving field powered by the newer sequencing technologies (collectively termed next-generation sequencing [ NGS ]). These technologies enable the sequencing of deoxyribonucleic acid (DNA) and ribonucleic acid much more quickly and cheaply than the previously used Sanger sequencing, and in concert with the ability to store and analyze huge data sets via computers have revolutionized the study of genomics and molecular biology and the direct clinical application of this knowledge. For example, whole exome sequencing and whole genome sequencing is increasingly being applied and is becoming part of established medical diagnostic practice in several situations, for example, neonates with multiple congenital malformations or metabolic disturbances in the newborn intensive care unit. Use of fetal DNA in maternal cell free plasma is becoming the standard for antenatal diagnosis of aneuploidy, without the need for invasive screening procedures, such as amniocentesis or chorionic villous sampling, and has been used to diagnose congenital adrenal hyperplasia in at-risk pregnancy of heterozygote carriers of this disease. NGS allows prompt genetic diagnosis of different mutations responsible for a specific disease or syndrome complex, for example, Neonatal Diabetes Mellitus, so that remedial treatment can be expeditiously provided, thereby altering what has hitherto been considered the natural history of the particular disorder. Direct therapeutic applications, such as, for example, choice of drug for maximum effectiveness, while avoiding drug interactions or excluding drug sensitivity, are also being explored. Gene therapy and gene editing for pediatric endocrine disorders are on the horizon and some show promise, but with many practical and ethical issues still to be resolved, as discussed in Chapters 2 and 3.
Unique aspects of pediatric endocrinology
Fetal Origins of Adult Disease
Pediatrics is all about growth and development of each aspect of human life—physical, emotional, cognitive, and sexual—from conception through birth, neonatal adaptation, infancy, childhood, puberty, and young adulthood. Changes in the mature organism continue, but the tempo is considerably slower than the rapid changes of early life. The endocrine system plays a central role in these adaptations and changes. It has long been known that environmental insults, such as viral infections (rubella, herpes, human immunodeficiency virus [HIV], cytomegalovirus, Zika) or drugs or other chemicals (thalidomide, phenytoin, alcohol), particularly in the first trimester, can result in distinct patterns of embryopathy. Only fairly recently has it been appreciated that developmental plasticity is influenced by the intrauterine nutritional environment that may predispose to the later development of diseases, such as type 2 diabetes. The notion that lack of nutrients during critical periods of development impart subsequent greater risk for development of adult diseases, such as hypertension, stroke, coronary disease, and diabetes emanated from careful epidemiologic observations of cohorts, such as those of the Dutch famine toward the end of the second world war and a British cohort of citizens with health records from birth to death. The former documented that the effects of the famine appeared to depend on its occurrence during gestation, early gestation being the most vulnerable period, with effects on schizophrenia, depression, atherogenic lipid profile, and with an increased rate of hypertension and coronary heart disease later in life. The latter demonstrated that a limited supply of nutrients, as reflected in intrauterine growth restriction, even in the absence of frank famine, could permanently alter the metabolism and other physiologic aspects in later adult life, including stroke and diabetes. Moreover, these effects could persist into future generations, suggesting modifications of the genome, giving rise to the concept of fetal origins of adult disease, as proposed by Barker. As a hypothesis, the “fetal origins of adult disease” is now well supported by epidemiologic and experimental data, including the epigenetic modification of gene expression, via patterns of methylation or other modifications, some affecting the expression of genes regulating insulin secretion, cortisol, and other hormones. These interactions are not restricted to early development—they may occur in the third trimester or beyond, but once “imprinted,” they may be passed on to succeeding generations. Thus the intrauterine and peripartum environment may permanently modify the expression of genes, including those of the endocrine system.
Acquisition of Patterns of Hormone Secretion and Action
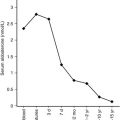
At any given time, the plasma concentration of a hormone reflects its synthesis, secretion, and clearance, a concept that implies removal from the circulation in units of time. Indeed, the relationship between the production rate (PR) of a hormone (units/time), the serum or plasma concentration (C = units/mL), and its metabolic clearance rate (MCR) from the plasma (mL/time) is defined by the formula PR = MCR × C, so knowing any two of these three variables permits one to solve the third. However, the concentration of a hormone may vary depending on such factors as time of day, stages of sleep, stage of puberty, renal or hepatic function, and the ingestion of other drugs. Hence each of these aspects represents complex interactions. For example, the secretion of ACTH and the chief product of its action, cortisol, are related in phase, the former preceding the latter and being highest in the early morning and lowest toward midnight. ACTH synthesis may have a basal rate determined by the impulses from its hypothalamic-releasing hormone, known as corticotropin-releasing hormone ( CRH ), but both display a diurnal rhythm entrained by the light/dark cycle and relayed to the suprachiasmatic nucleus of the hypothalamus. As the fetus is not exposed to light/dark cycles and the newborn spends a majority of time asleep, it is important to know when the diurnal rhythm becomes established. This has importance for undertaking measurements of cortisol in the newborn to determine if the hormone is deficient, as, for example, in evaluating newborn hypoglycemia. Also, stress results in a rapid increase in the CRH-ACTH-cortisol axis and the secretion of cortisol. Such rapid adaptation is essential for the appropriate adjustments to physiologic stress resulting in the “fight or flight response.” It is known that the hypothalamic pituitary adrenal axis is established in utero and functional because inborn errors of metabolism, such as those found in congenital adrenal hyperplasia as a result of defects in enzymes responsible for cortisol synthesis, result in markedly elevated ACTH in utero, with hypertrophy of the adrenal cortex. However, after birth, there is a brief period during which adrenal secretion of cortisol is low, as reflected in the plasma levels, and it is not known how rapidly the newborn can adjust to a stressful situation by appropriate increments in the ACTH-cortisol secretion rates. Some hormonal cycles, such as GH secretion and FSH/LH secretion at the onset of puberty, are related to sleep rather than the light/dark cycle, as discussed for GH 5 and separately for the gonadotropins in the relevant chapters of this book. But it is not known if these sleep-regulated patterns are operative in the newborn. Following birth, some inhibitory pathways of hormone secretion are not yet established, resulting in high hormone concentrations whose functional significance is not known. For example, inhibition of GH secretion by somatotropin release-inhibiting factor appears to develop only after birth, so that GH concentrations in serum are quite high in the newborn (averaging approximately 40 ng/mL), values that would be consistent with acromegaly in an adult. On the other hand, the expression of GH receptors on tissues and their linkage to postreceptor events is delayed and become fully operational only after several months, so that the effects of the high GH concentrations are muted. The practical implications of these findings are that GH deficiency in a full-term newborn will not be characterized by discernible small size at birth. Indeed, GH deficiency does not become manifest in a delayed growth velocity, until after 3 to 6 months when the GH–growth hormone receptor axis becomes established. Yet GH plays an important role in maintaining glucose homeostasis in the newborn, because GH deficiency may be associated with hypoglycemia, and this deficiency can be diagnosed without the need for stimulation tests if the GH concentration is lower than 10 ng/mL in the first week of life of a full-term newborn. Another hormone whose concentrations remain very high at birth and for several weeks thereafter is prolactin, presumably because the neurologic pathways responsible for dopamine secretion are not yet fully developed. The physiologic implications of these adaptive processes in the secretion and action of prolactin in the newborn are not fully understood.
Adaptations in Endocrine Function at Birth
Separation of the newborn baby from its maternal blood supply after birth imposes a near-instantaneous need for adaptations in functions, such as the requirements of oxygen, maintenance of body temperature, and sources of nutrients. The endocrine system plays a vital role in several of these adaptations; three are briefly described here and are explored in more detail in relevant chapters in this book.
Maintenance of Body Temperature
The hypothalamic-pituitary-thyroid axis is intimately involved in the adaptations of body temperature regulation. The fetus, bathed in amniotic fluid and supplied by maternal blood, maintains a temperature of 37° C in utero. Delivery into an ambient temperature of approximately 20° to 25° C in a modern delivery room represents a significant fall in ambient temperature that contributes to activation of thyroid function. The concentration of TSH increases approximately 10-fold between birth and 15 to 30 minutes after cutting of the umbilical cord, to values of approximately 100 μU/mL. Simultaneously, type 2 deiodinase is activated, converting thyroxine (T 4 ) to triiodothyronine (T 3 ) rather than the fetal pattern of type 3 deiodinase that converts T 4 to reverse T 3 . These rapid changes result in a surge of T 3 concentration, a decline in reverse T 3 concentration, and a somewhat later rise of T 4 concentration in the newborn’s blood. Together, these coordinated changes in thyroid function enable T 3 to act on brown adipose tissue to activate nonshivering thermogenesis. Although TSH concentrations decline to values below 10 μU/mL by day 2 to day 3 of life, T 3 and T 4 values remain elevated for days to weeks at concentrations that would be consistent with thyrotoxicosis in older children and adults (see Chapter 8 for details). Hence values of thyroid function reported for full-term or premature newborns must be reported as age specific; they may often be labeled erroneously as hyperthyroid in laboratories that only list reference values suitable for adults. Not only do premature babies have thyroid hormone–related concentrations that are lower than those of full term infants, but the acute stresses of prematurity, such as sepsis or respiratory distress syndrome, may lead to the “euthyroid sick syndrome” and further complicate the interpretation of thyroid function tests, as discussed in Chapter 8 .
Glucose Homeostasis
The fetus obtains all of its glucose via placental transfer from the mother, with little, if any, endogenous glucose production until delivery. Following cord cutting, epinephrine and glucagon each rise approximately three- to five-fold, growth hormone is high at approximately 40 ng/dL (as mentioned previously), as are cortisol values, which are highest at about 2 hours after birth and, on average, remain in the range of 2.7 to 7.6 μg/dL, mostly in the free form, in the first week of life. The coordinated effects of these four classic “counterregulatory” hormones, together with a small fall in insulin, stem the initial decline in blood glucose concentration, activate glycogen breakdown and gluconeogenesis, and initiate lipolysis with later activation of ketone body production by day 2 to day 3 of life. Understanding these critical adaptations is essential for the appropriate management of hypoglycemia in a newborn, as detailed in Chapter 7 . All hormonal measurements must be age specific; a common problem with hypoglycemia in the newborn is the range of normal fasting insulin values from an adult reference range, generally in the range of 5 to 29 μU/mL. A newborn with hypoglycemia who at the time of hypoglycemia has an insulin value greater than 5 μU/mL likely has some form of genetic or acquired hyperinsulinism.
Gonadotropins and Sex Hormones
In males, testosterone concentrations on day 1 of life are high, ranging from 75 to 400 ng/dL, values that are consistent with Tanner stages 3 to 4 of male puberty. These high concentrations decline rapidly within the first few days after delivery but remain elevated at 20 to 50 ng/dL compared with males aged 1 to 10 years, in whom values are less than 10 ng/dL. A second rise in testosterone concentrations occurs between 1 week and 1 to 2 months, the mean values being approximately 200 ng/dL. Both FSH and LH also are relatively high in males at this age of life and decline to prepubertal levels only at about the end of the first year. In females, concentrations of estradiol are markedly elevated after birth and fall rapidly during the first week of life to prepubertal values, with a secondary rise occurring between 30 and 60 days followed by decline to prepubertal concentrations after 1 to 2 years. Values of FSH may range up to 14 mIU/mL in females and decline more slowly than in males, reaching prepubertal values only after 2 to 3 years. Likewise, LH values in females may be in the classical pubertal range in the first few months of life and decline to prepubertal values only after 1 to 2 years. This so called minipuberty of infancy is discussed in greater detail in Chapters Chapter 6 , Chapter 16 , and Chapter 18 . A precise function for these perinatal changes in sex hormones and gonadotropins is unknown, but it has been proposed that they may have relevance to developmental patterning of male or female neural function. In addition, clinical relevance is related to the common problem of thelarche in newborn-infant females, as discussed in Chapter 16 .
In summary, each of the major endocrine organs and systems undergo dramatic neonatal adaptations, so that knowledge of these endocrine adaptations following birth is essential for the appropriate evaluation of suspected abnormalities in endocrine function, and the interpretation of age- and sex-specific values of circulating hormones or other metabolic variables. A major deficit in healthcare systems is the application of standards of normal ranges for hormones or other biochemical indices derived from adult reference ranges to the newborn and infant; values must be relevant to the age- and sex-specific situation being evaluated.
Evaluating endocrine disorders in infancy and childhood
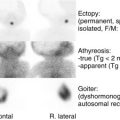
A general principle of pediatric endocrinology is that the earlier the manifestation of either underactivity or excess hormone function, the more likely the cause is to be a genetic disorder, together with possible structural abnormalities. For example, the entity of septooptic dysplasia with underactivity of anterior and/or posterior pituitary function may be associated with typical structural abnormalities of optic nerve hypoplasia, absence of the septum pellucidum or corpus callosum, a small anterior pituitary, interruption or absence of the pituitary stalk, and an ectopic or absent posterior pituitary bright spot on magnetic resonance imaging (MRI) of the brain. In the majority of these patients, no genetic abnormality can be detected. However, mutations in three genes ( HESX1 , OTX2 , and SOX2 ) that play a role in the embryonic development of the pituitary gland, eyes, and forebrain, have been identified in a small subset of these patients. Hypothyroidism found on newborn screening is most commonly associated with an ectopic thyroid gland; total absence or goitrous hypothyroidism should suggest a defect in the genes responsible for thyroid gland formation ( TTF-1 , TTF-2 , PAX8 ) or an “inborn error of thyroid hormone synthesis” (see Chapter 8 ). To be sure, perinatal events and maternal illness or medication must be considered. For example, perinatal asphyxia or a difficult delivery may be associated with later evidence of hypopituitarism. Maternal ingestion of antithyroid medications would result in their transfer across the placenta and may cause transient neonatal hypothyroidism, as these agents affect the fetal thyroid in the same way as they affect the maternal thyroid gland. Similarly, transfer of immunoglobulin G antibodies that block or stimulate thyroid function will result in newborn hypothyroidism or hyperthyroidism that lasts for several weeks, until maternal antibodies are cleared from the circulation. A newborn with severe hyperthyroidism in the absence of any evidence of autoimmune disease in the mother almost certainly has an activating mutation of the TSH receptor (see Chapter 8 ).
More subtle defects in endocrine function may appear later in childhood, but still have a genetic basis, for example, hypogonadotropic hypogonadism. This entity may be considered at birth in male infants because of underdeveloped penile and testicular size, but often does not become manifest or discovered until delayed puberty is investigated. Macrosomia in an infant born to a mother with poorly controlled diabetes mellitus reflects secondary hyperinsulinism in the fetus, with hypoglycemia in the newborn when the maternal supply of glucose is curtailed. However, similar features in a baby born to a healthy young mother should immediately suggest the possibility of a genetic form of hyperinsulinism, most commonly the result of an inactivating mutation in the genes regulating the adenosine triphosphate (ATP)–regulated potassium channel (K ATP ). If the biochemical profile and clinical course are consistent with hyperinsulinism, but insulin concentrations are low, consider an activating mutation in the insulin receptor signaling pathway. Conversely, significant intrauterine growth retardation may be caused by many factors and is a major feature of neonatal diabetes mellitus, so early diagnosis via measurement of glucose may be lifesaving by providing insulin or sulfonylurea drugs, to which some 90% of newborns with activating mutations of the KCNJ11 gene respond with improved endogenous insulin secretion and restoration of near normal glucose metabolism and as discussed in detail in Chapter 10 . Severe intrauterine growth retardation also is a feature of inactivating mutations in insulin receptors, for example, Donohue syndrome or Rabson-Mendenhall syndrome. Defects in the secretion or action of IGF-1 mimic the effects of insulin secretion or action, both hormones and both receptors being structurally, as well as functionally related.
Autoimmunity, trauma, and chemo/radiotherapy for a childhood malignancy are the most common causes of acquired endocrine disorders. Thus as in all of medicine, a careful and thorough history, careful physical examination, and targeted investigation of hormonal measurements (taking into consideration age, time of day, and the value of obtaining a “free” hormone versus total hormone measurement) form the basis of the diagnostic approach. This may be followed by imaging of the suspected organ involved to diagnose the basis of the suspected endocrine dysfunction, in the particular patient being investigated. Increasingly, molecular diagnostics is becoming an integral component of this evaluation, both to establish the cause of the entity and to provide guidance for prognosis, such as multiple endocrine neoplasia 2 (see Chapter 15 ).
Definitive diagnosis may require stimulation tests (e.g., for growth hormone deficiency or with ACTH for suspected adrenal insufficiency) because a single random value is not sufficiently informative. Such stimulation tests are an integral and essential component of diagnosis and management of endocrine disorders at any age but particularly in the pediatric age range, where performance of the test must take into consideration its safety, easing anxiety and correct sample handling.
Finally, modern biology has provided pure synthetic compounds to replace what may be missing, such as thyroid hormone or cortisol, administration of pulsatile gonadotropin-releasing-hormone (GnRH) for syndromes of hypogonadotropic hypogonadism via programmable pumps, GnRH analogues for suppressing puberty, growth hormone for GH deficiency or other forms of approved short stature, various insulin preparations with ultrashort, short, intermediate, long-acting and ultralong acting forms for managing diabetes, and ultralong-acting somatostatin for childhood acromegaly as occurs in Carney complex, or some patients with McCune-Albright syndrome. Hormone replacement must also take into consideration administration via the dermal rather than the oral route to avoid “first pass” considerations in bypassing the liver, as, for example, giving sex hormones, such as estrogen to females with delayed puberty, including Turner syndrome (see Chapters 16 and 17 ).
Concluding remarks
Endocrinology is the science of cellular communication that enables the biochemical integration of life’s vital processes. Pediatric endocrinology is the linchpin for these processes during the developmental epoch from fetus to mature adult. Evolving developments in molecular biology, bioinformatics, pharmacogenetics, and bioimaging will ensure that this specialty remains at the forefront of pediatric research and practice. This chapter is intended as an introduction to this important field, with greater detail to be found in the chapters that follow. Much has been learned since the prior edition of this chapter in 2014 and some of this newer knowledge is incorporated here; much remains to be learned.
References
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


