Chapter Outline
CLINICAL PRESENTATION AND EVALUATION
Osteosarcoma is the most common primary bone tumor. There are approximately 600 cases/year in the United States. The term osteosarcoma was first used in the early 1800s by Alexis Boyer, the imperial family surgeon for Napoleon. Although low-grade forms of osteosarcoma do exist, more than 90% of osteosarcomas are high-grade malignant lesions. Low-grade osteosarcomas will be discussed, but most of what follows pertains to high-grade osteosarcomas.
Osteosarcoma usually occurs in adolescents and young adults. Presenting symptoms can include tumor-related pain with or without a mass, a painless mass, and a pathologic fracture. The distal femur is the most frequent site of primary disease, followed by the proximal tibia and then the proximal femur, hip, and proximal humerus. As early as 1818 it was recognized that untreated patients with osteosarcoma often developed metastatic disease. The most common site of metastasis is the lungs, followed by bone. Metastatic disease is present in approximately 15% of patients at the time of diagnosis.
Prior to the 1970s, when chemotherapy was first used to treat osteosarcoma, overall survival (OS) was approximately 20%. With modern chemotherapy and surgical control, the 10-year OS of patients with localized osteosarcoma is around 70%. The OS is much worse for patients with metastases at diagnosis and for those with relapsed disease. In both groups, the 5-year OS rate is approximately 25%. The most active chemotherapy agents in osteosarcoma are doxorubicin (Doxo), cisplatin (CP), and high-dose methotrexate (HDMTX). Ifosfamide and etoposide are also active against osteosarcoma and are typically used in addition to Doxo, CP, and HDMTX in patients with metastatic disease and in patients with recurrence.
In the past three decades, a number of discoveries have led to a better understanding of osteosarcoma biology. For example, the tumor suppressor gene TP53 and the retinoblastoma gene RB1 are altered in osteosarcoma. Oncogenes, including MYC, MET, and FOS, have also been implicated in osteosarcomagenesis. These tumor suppressors, oncogenes, and other biologic factors will be discussed in detail. Unlike most childhood cancers, there has been minimal improvement in OS over the past three decades. Consequently, one of the research priorities in osteosarcoma is the identification of additional active therapies.
Epidemiology
Osteosarcoma is the most common primary bone tumor in children. The average annual incidence of osteosarcoma in children younger than 20 years in the United States is 4.8/million. Approximately 3% of all malignancies in this age group are osteosarcomas. In 2000, the most recent year for which data are available, there were 440 cases of osteosarcoma in children from birth to 19 years of age in the United States. There were an additional 135 cases in young adults aged 20 to 29. Internationally, incidence rates approximate the U.S. rate, with the exception of a higher incidence in some African countries. In Sudan and Uganda, the osteosarcoma incidence in children younger than 14 years is 5.3 and 6.4/million respectively, compared with an incidence of between 2 and 3/million in this age group in the United States, Europe, and Asia.
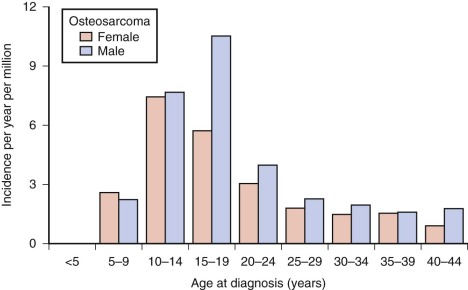
Osteosarcoma occurs rarely in children younger than 5 years. After age 5, the incidence increases steadily, reaching a peak at age 15 years. After the adolescent peak, the rate of osteosarcoma steadily declines, leveling off at a rate of 1 to 2/million. A second peak in incidence, approximately half the magnitude of the adolescent peak, occurs in the sixth to seventh decade. Osteosarcoma in older patients is associated with Paget disease and prior radiation therapy, although approximately half of older patients with osteosarcoma have neither condition. The adolescent peak occurs at age 13 years in girls and between ages 15 and 17 years in boys ( Fig. 62-1 ). The age at peak incidence corresponds to the age of greatest growth velocity in each gender. This association contributes to the evidence supporting a role for growth in the cause of osteosarcoma (see “ Etiology ”). Osteosarcoma is slightly more common in males, particularly in the 15- to 19-year-old age group.
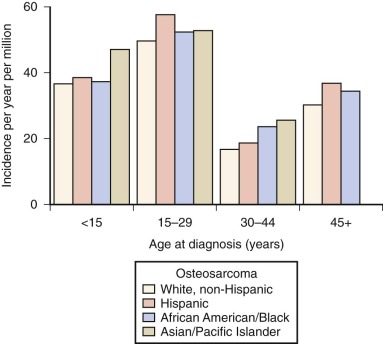
In the United States, osteosarcoma is slightly more common in African Americans, Hispanics, and Asians and Pacific Islanders ( Fig. 62-2 ). Several studies have reported that rates of osteosarcoma in black children younger than 14 years are about twice those in white children of the same age. In the most recent report on osteosarcoma from the Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute, U.S. blacks younger than 20 years continue to have a slightly higher rate of osteosarcoma. As shown in Figure 62-2 , for the age group 15 to 29 years, the racial group with the highest rate of osteosarcoma is Hispanic. The relative risk of developing osteosarcoma for Hispanic children younger than 14 years of age, based on data collected between 1975 and 2003, is 1.3. Asians and Pacific Islanders have the highest rate of osteosarcoma in the age groups younger than 15 years and 30 to 44 years. However, the reported incidence in Hispanics and in Asians and Pacific Islanders is likely to be inaccurate given the small absolute number of affected individuals and uncertainty in the denominator (i.e., the number of Hispanics and Asians and Pacific Islanders within each age group living in the United States). In summary, there is evidence for a slightly increased incidence of osteosarcoma among U.S. blacks and Hispanics. Recent SEER data show an increased rate in Asians and Pacific Islanders younger than 15 years, but this finding needs further investigation.
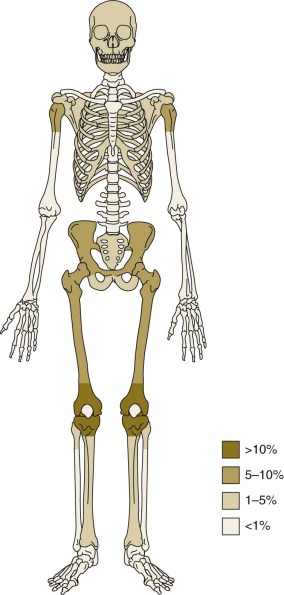
The most common anatomic sites of osteosarcoma at initial presentation are the long bones of the lower limbs. Fifty percent of osteosarcomas are located around the knee, in the distal femur, proximal tibia, or proximal fibula ( Fig. 62-3 ). Approximately 10% of tumors occur in the mid or proximal femur, and 9% occur in the proximal humerus. Of patients with osteosarcoma, 10% to 20% have metastatic disease at initial presentation. The most common site of metastasis is the lungs. Isolated pulmonary metastases are present in 61% of patients with metastatic disease at presentation. Isolated metastases to bone are present in 16% of patients, and 14% have both pulmonary and bone metastases. Seven percent of patients have metastases to other rare sites plus bone and/or pulmonary metastases, and 2% of patients have isolated metastases to rare sites. Rare sites of metastasis in patients with primary metastatic disease, as opposed to metastatic disease in patients with recurrent disease, are lymph nodes, central nervous system, liver, adrenal gland, and soft tissues.
Etiology
When a patient presents with osteosarcoma, it is often difficult to identify a cause. However, recognition of causative factors has led to the discovery of biologic mechanisms of transformation, as in the case of predisposition to osteosarcoma in Li-Fraumeni syndrome, or the identification of exposures to be avoided, as in the case of radiation-induced osteosarcoma. Inherited syndromes in which there is an increased occurrence of osteosarcoma are hereditary retinoblastoma and Li-Fraumeni, Rothmund-Thomson, Bloom, and Werner syndromes. There is a clear association between the risk for osteosarcoma and prior radiation therapy or treatment with alkylating chemotherapy. Epidemiologic studies and clinical observation have suggested that increased height may be a risk factor for the development of osteosarcoma. Finally, environmental exposures to radium and beryllium likely increase the risk for osteosarcoma, but exposure to high doses of these agents is rare.
As noted, peak rates of osteosarcoma coincide with the period of maximal linear growth during puberty. Most osteosarcomas occurring in the adolescent age group are in the long bones of the axial skeleton, the bones in which the most active growth occurs. These clinical observations have led to the hypothesis that the hormonal milieu during maximal linear growth encourages the development of osteosarcoma and that, by extension, osteosarcoma would be more common in taller adolescents. Multiple epidemiologic studies have addressed this question, but results are contradictory. Approximately 50% of the studies have found that osteosarcoma patients are taller at diagnosis than age-matched controls or the normal population. The remaining studies have not supported an association between height and the occurrence of osteosarcoma. The larger studies and those with more reliable sources of height data have shown a positive association between tall stature and osteosarcoma. These data, in part, have led to the investigation of the insulin-like growth factor axis in osteosarcoma (see “ Biology ”).
Approximately 1% of patients develop secondary bone tumors after treatment of a primary pediatric malignancy ; of these secondary bone tumors, 50% to 80% are osteosarcomas. On the other hand, secondary Ewing sarcoma is rare. The relative risk of secondary osteosarcoma increases greatly with radiation doses to the bone higher than 1000 cGy. In patients who received between 3000 and 5000 cGy to the bone during treatment of their primary pediatric malignancy, the relative risk of secondary osteosarcoma was variably reported, but it may be as high as 100. A study in patients with primary Ewing sarcoma has concluded that only higher radiation doses increase the risk for secondary sarcomas, with no tumors developing in patients who had received less than 4800 cGy. The highest risk, 130 cases/10,000 person-years of observation, was observed among patients who had received 6000 cGy or more. In the 92 published cases of secondary osteosarcoma for whom radiation dose was reported, the median radiation dose delivered was 4500 to 5400 cGy. The average time between diagnosis of the primary malignancy and development of secondary osteosarcoma is 10 years. Another factor that increases the likelihood that survivors of a primary cancer will develop secondary osteosarcoma is treatment with alkylating agents. This class of chemotherapy increases the relative risk of secondary osteosarcoma by a factor of 5.
Data regarding an increased risk for cancer in people exposed to low levels of radiation from the atomic bombs in Hiroshima and Nagasaki have led to increased concern about the risk of diagnostic imaging with x-rays and computed tomographic (CT) scans, particularly in pediatric patients. However, current evidence does not suggest that osteosarcoma risk would be increased from commonly used imaging modalities. In particular, the radiation dose of a CT scan is approximately 6 cGy and the risk for bone cancer in pediatric cancer survivors who received less than 1000 cGy to the bone is not increased.
Osteosarcoma is seen in a number of inherited cancer predisposition syndromes. These syndromes have often illuminated mechanisms of transformation in sporadic osteosarcoma. Children with hereditary retinoblastoma have a germline heterozygous, inactivating deletion, and/or a frameshift or point mutation of the tumor suppressor gene retinoblastoma 1 (RB1). Affected children develop retinoblastoma usually in one or both eyes. The cumulative incidence of a nonretinoblastoma malignancy in hereditary retinoblastoma is 51%, and approximately 40% of these second malignancies are osteosarcomas. Both osteosarcomas and retinoblastomas in these patients have loss of heterozygosity of the RB1 allele, resulting in a complete loss of RB1 function. As discussed later (see “ Biology ”), loss of RB1 function also occurs in sporadic osteosarcomas.
In the Li-Fraumeni syndrome, patients carry a heterozygous inactivating mutation of the tumor suppressor gene TP53. Affected individuals have a greatly increased cancer risk at a young age. The most common malignancies are breast cancer and bone and soft tissue sarcomas, brain tumors, and leukemia. Slightly more than 10% of the malignancies in patients with Li-Fraumeni syndrome are osteosarcomas. Among patients with Li-Fraumeni syndrome, mutations in the TP53 DNA-binding domain (as compared with mutations outside the DNA-binding domain or null mutations) are associated with early sarcomas. Patients with osteosarcomas and other cancers that develop in Li-Fraumeni syndrome have loss of the normal TP53 allele consistent with the tumor suppressor function of the TP53 gene product. Loss of TP53 function by TP53 mutation or amplification of MDM2 or COPS3 is also present in most sporadic osteosarcomas (see “ Biology ”).
Rothmund-Thomson, Bloom, and Werner syndromes are members of a family of autosomal recessive disorders caused by mutations in one of the RECQ DNA helicases. Rothmund-Thomson syndrome, caused by mutations in the RECQL4 gene, is characterized by rash, skeletal dysplasias, and sparse hair. Up to 30% of patients with Rothmund-Thomson syndrome develop osteosarcoma at a slightly younger age than in sporadic osteosarcoma. Bloom syndrome, caused by mutations in the BLM (RECQL2) gene, is characterized by short stature, telangiectasia, and an increased risk for developing a wide variety of cancers at a young age. Unlike in Rothmund-Thomson syndrome, only a small percentage of cancers occurring in affected individuals are osteosarcomas. Werner syndrome is caused by a mutation in the WRN (RECQL3) gene. Patients have scleroderma-like skin changes, premature aging, and a typical body habitus with short stature, long spindly limbs, and a stocky trunk. Like in Bloom syndrome, the risk for developing cancer at a young age is increased in Werner syndrome. Also like in Bloom syndrome, osteosarcoma constitutes fewer than 10% of the malignancies occurring in patients with Werner syndrome. For a further discussion of the role of RECQ DNA helicases in osteosarcoma, see “ Biology ”.
The risk for developing osteosarcoma is increased in adults with Paget disease. In Paget disease, which affects up to 1% of adults older than the age of 40 years, bone resorption by osteoclasts is increased. As a result, there is a compensatory increase in osteoblast activity and bone formation. Osteosarcoma develops in less than 1% of patients with Paget disease, but this represents a higher rate than that seen in the general population. The average age of patients with Paget disease–related osteosarcoma is 70 years.
A single genome-wide association study has been conducted in osteosarcoma. There are two susceptibility loci achieving statistical significance in osteosarcoma. One locus occurs in a gene desert and the other, at 6p21.3, implicates GRM4, a glutamate receptor gene involved in inhibition of the cyclic adenosine monophosphate (AMP) cascade.
Previously, some believed that trauma may cause osteosarcoma. However, modern epidemiologic studies have not supported a relationship between trauma and osteosarcoma. Prior observations linking trauma and osteosarcoma were likely the result of the fact that a traumatic event may result in a previously existing osteosarcoma being brought to medical attention.
Environmental exposures, other than ionizing radiation, for which there is evidence of an association between exposure and osteosarcoma risk, include exposure to radium and beryllium. Radium is a radioactive element that is variably present in well water. When ingested, radium retained in the body is deposited in bone. Studies of those exposed to high levels of radium while painting dials on clocks in the 1920s strongly indicated that exposure to high doses of radium causes osteosarcoma. The risk for osteosarcoma from exposure to low levels of radium in drinking water is less clear. Two studies have shown a slight increase in risk for bone sarcoma or osteosarcoma in those born in areas with drinking water containing relatively high concentrations of radium. Conversely, two studies have shown no link between mean levels of radium in water and osteosarcoma. Beryllium is a metallic element with limited use in the production of aerospace structural material, x-ray tubes, and nuclear reactors. Although intravenous injection of beryllium salts causes osteosarcoma in rabbits, there is limited evidence of an increased risk for osteosarcoma caused by beryllium exposure in humans. Case-control studies have failed to identify other environmental or occupational exposures that increase the risk for osteosarcoma. Because fluoride can stimulate bone formation there has been concern about a possible relationship between fluoride in drinking water and osteosarcoma. This is a very difficult question to study adequately given that multiple factors beyond water fluoridation may impact fluoride intake. So far, there is no substantial evidence to support a causative association between fluoride exposure and osteosarcoma.
Clinical Presentation and Evaluation
Clinical, laboratory, and radiologic findings are often highly suggestive of osteosarcoma but are not pathognomonic. Biopsy is always required for diagnosis. Atypical imaging characteristics, including a benign appearance in high-grade osteosarcoma, have been described. The differential diagnosis of osteosarcoma includes benign and malignant bone tumors ( Box 62-1 ; Fig. 62-4 ).
Other Malignant Primary Bone Tumors
Ewing sarcoma
Chondrosarcoma
Fibrosarcoma
Langerhans cell histiocytosis
Other Malignancies Presenting as Bone Tumor(S)
Lymphoma
Neuroblastoma
Metastatic rhabdomyosarcoma
Metastatic melanoma
Benign Bone Tumors
Aneurysmal bone cyst
Osteoblastoma
Osteoid osteoma
Giant cell tumor
Unicameral bone cyst
Infection
Osteomyelitis
Clinical Manifestations
Clinical symptoms of osteosarcoma are nonspecific; 25% of patients have pain and a palpable mass at presentation, and 70% of patients present with pain as their only complaint. Pain tends to be intermittent and exacerbated by activity. The remaining patients present with a painless mass. The duration of symptoms before diagnosis ranges from 1 day to 5 years, with a median of about ![]() months. In approximately 10% of patients, symptom duration is longer than 6 months. A pathologic fracture is present in 8% of patients at the time of diagnosis.
months. In approximately 10% of patients, symptom duration is longer than 6 months. A pathologic fracture is present in 8% of patients at the time of diagnosis.
Radiologic Evaluation
Radiologic evaluation in osteosarcoma informs the initial diagnostic impression, aids in the determination of the best approach for biopsy, facilitates surgical resection, and identifies sites of metastatic disease, if present. The recommended imaging studies for osteosarcoma at the time of diagnosis are plain radiography and magnetic resonance imaging (MRI) of the primary tumor site to evaluate the extent of local disease, a bone scan to screen for bony metastases, and a CT scan of the chest to determine whether pulmonary metastases are present. Whenever possible, these imaging studies should be performed before diagnostic biopsy because postsurgical atelectasis interferes with evaluation of the lung parenchyma. MRI of the primary tumor should include the entire bone from which the tumor arises and the closest adjacent joint to screen for skip metastases properly. An additional MR image should be obtained after neoadjuvant chemotherapy has been given but before definitive resection to determine the best plan for the surgical approach.
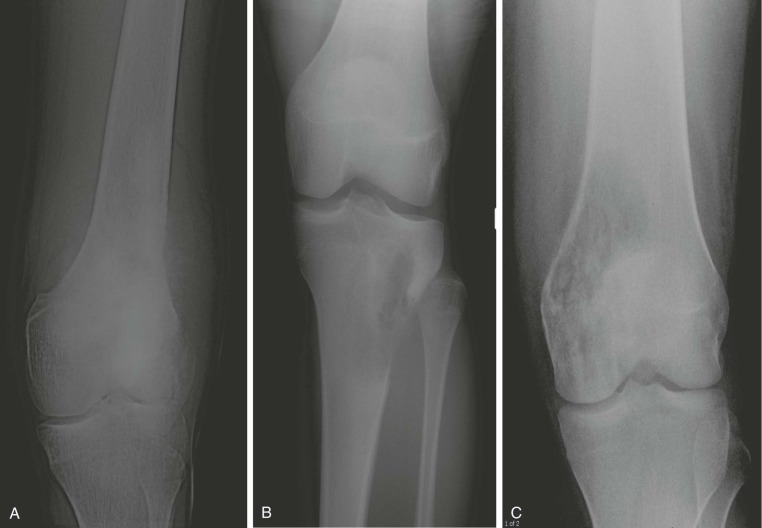
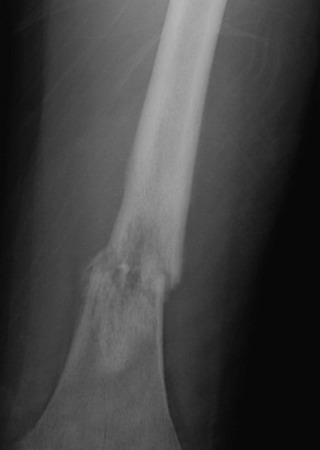
A plain radiograph of the primary tumor has usually been obtained before referral to an oncologist or orthopedist. Most appendicular osteosarcomas are located in the metaphyseal portion of the long bone. Both lytic destruction of bone and sclerotic irregular new bone formation can occur. Osteosarcoma usually has a mixed pattern of bone lysis and sclerosis, but one of these patterns can predominate. Regardless of the predominant pattern, the boundary between tumor and normal bone is usually irregular. The bone cortex is often disrupted by tumor growth into the surrounding soft tissue. Osteoid formation within the tumor results in areas of calcification visible in the bony and soft tissue components of the tumor. Periosteal new bone formation in osteosarcoma can be irregular or can occur in a sunburst pattern. “Sunburst” refers to linear new bone growth that is perpendicular to the bony cortex and arranged in a fanlike pattern ( Fig. 62-5 ). In contrast, in Ewing sarcoma, periosteal new bone growth typically occurs in an onionskin pattern in which linear new bone is oriented parallel to the bony cortex. Periosteal new bone formation in osteosarcoma can also be present at the edge of the preserved cortex, resulting in the Codman triangle. Plain radiographs also assist in the diagnosis of pathologic fractures ( Fig. 62-6 ).
Although plain radiographs of the primary tumor are informative, MRI is preferable for local staging, presurgical planning, and identification of skip metastases. CT is an acceptable alternative to MRI, but most orthopedic surgeons and oncologists prefer MRI. In two studies directly comparing preoperative CT and MRI results with pathologic findings in patients with osteosarcoma, MRI was found to be superior to CT in defining the longitudinal extent of intraosseous tumor, extension into adjacent muscle compartments, and involvement of the neurovascular bundle. The two modalities were equivalent in their ability to define cortical bone and joint involvement. However, a more recent study in a larger patient population did not find a statistically significant difference between CT and MRI in their ability to predict the involvement of bone, muscle, joints, and the neurovascular bundle. One possible explanation for the contradictory results is improvements in CT quality during the interval between the studies. In addition, the later study grouped all primary bone tumors together for analysis but, given that 60% of the bone tumors were osteosarcoma, results would likely be similar for osteosarcomas alone. In comparison to CT and technetium-99 m ( 99m Tc) bone scanning, MRI more accurately predicts the intraosseous extent of osteosarcoma. The presence of significant edema has been associated with inaccurate assessment of the margins of tumor on MRI. Because edema can resolve with chemotherapy, imaging performed for the purpose of surgical planning should be obtained after the administration of neoadjuvant chemotherapy.
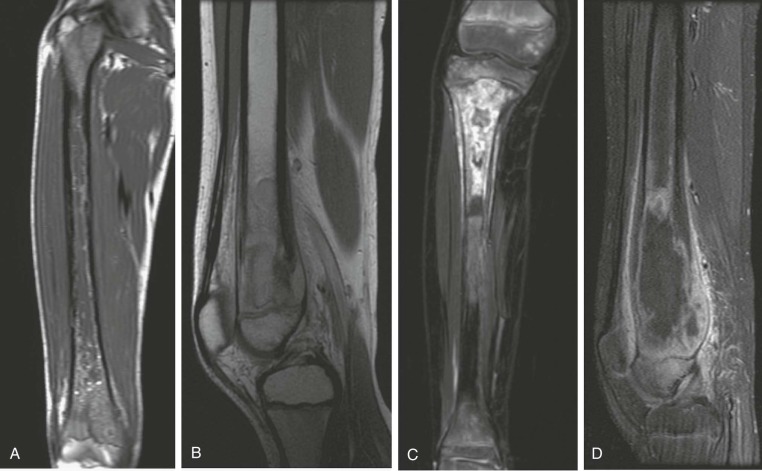
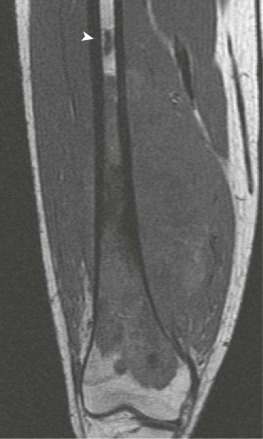
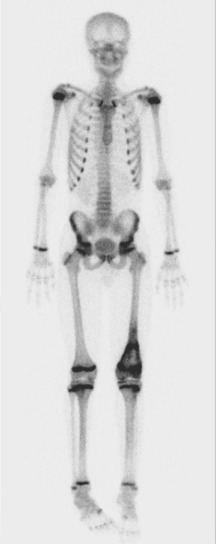
A skip metastasis is defined as a smaller distinct focus of osteosarcoma that is either within the same bone as the primary tumor or on the other side of the joint adjacent to the primary tumor. There must be normal structures not involved with tumor present between the primary tumor and skip metastasis. Skip metastases are synchronous with the primary tumor and thus are present at initial presentation. Diagnosis of skip metastases is of critical importance for prognostication and because complete surgical remission in osteosarcoma with skip metastases requires complete resection of both the primary tumor and skip metastases. Over 80% of skip metastases are visible on MR images ( Fig. 62-7 ). In contrast, skip metastases are visible on bone scan, CT scans, and plain radiographs in only 46%, 45%, and 36% of cases, respectively. Interestingly, a common reason for failure to identify skip metastases by bone scanning is merging of the signal from the primary tumor with the signal from the skip metastasis. To ensure that skip metastases are identified, MRI of the primary tumor should include the entire bone and adjacent joint. Bony metastases that are not skip metastases are best identified with a 99m Tc bone scan ( Fig. 62-8 ). The usefulness of 18 F-labeled fluorodeoxyglucose positron emission tomography (FDG-PET) in osteosarcoma has not yet been adequately evaluated. Results of preliminary studies have suggested that FDG-PET is more useful for assessing tumor response to chemotherapy and for posttherapeutic monitoring for recurrence than for assessing the initial tumor. Because edema can resolve with chemotherapy, imaging performed for the purpose of surgical planning should be performed after the administration of neoadjuvant chemotherapy.
CT of the chest is the most sensitive radiologic study for detection of pulmonary metastases. A generally accepted definition of definitive pulmonary metastatic disease is three or more lesions 5 mm or larger or one lesion 1 cm or larger. The interpretation of very small lesions (<5 mm) and solitary lesions between 5 mm and 1 cm detected by high-resolution CT can be difficult. In some cases, it is not possible to determine whether such CT findings represent metastatic disease or a nonmalignant lung process, and biopsy may be needed. Compared with a CT scan, a plain radiograph of the chest has a sensitivity of 57% and a 99m Tc bone scan has a sensitivity of 41%. Although CT is highly sensitive, evaluation at the time of thoracotomy usually reveals more pulmonary metastases than are appreciated on the CT scan. Importantly, chest CT scans should be obtained before biopsy of the primary tumor because postanesthesia atelectasis makes chest CT scans more difficult to interpret.
Whole-body MRI screening as a method of surveillance in patients with cancer predisposition is currently being evaluated. In a small group or individuals (n = 25) with hereditary retinoblastoma, whole-body MRI detected two cases of osteosarcoma. An additional individual was diagnosed with osteosarcoma due to symptoms. A screening protocol including whole-body MRI utilized in individuals with Li-Fraumeni syndrome detected no cases of osteosarcoma but did detect one case of malignant fibrous histiocytoma.
Biopsy
Traditionally, bone tumor tissue for pathologic examination was obtained by open biopsy. However, percutaneous core needle biopsies have become more common as the availability and sophistication of interventional radiology services have increased. A number of retrospective series have reported on the diagnostic yield and accuracy of core needle biopsies in osteosarcoma. Although none specifically studies pediatric osteosarcoma, all series that provide patient age included children. In these published series, core needle biopsy resulted in a definitive, correct diagnosis in 78% to 94% of cases. The more recently published series have reported a diagnostic accuracy of 90% or greater. However, even those percutaneous biopsies that yield sufficient tumor material for a diagnosis often do not yield sufficient tumor specimen for other biologic studies.
The outcome of percutaneous biopsy varies depending on the provider, technique, and patient population. In the published series reporting a high diagnostic yield, biopsies were performed by experienced interventional radiologists in consultation with orthopedic surgeons in tertiary or referral centers with considerable expertise in the diagnosis and management of bone tumors. In the more recent series, patients underwent conscious sedation or were given general anesthesia. Specific techniques included the use of fluoroscopic, CT, or ultrasound guidance and use of at least a 14-gauge needle. The tumor component targeted for biopsy was the soft tissue mass, if present, followed by the lytic aspect of an intraosseous lesion.
The most common reason for failure to obtain diagnostic tissue by core needle biopsy is the presence of a highly sclerotic tumor. Of the various types of osteosarcoma, telangiectatic osteosarcoma most often results in a nondiagnostic specimen or is misdiagnosed. Development of osteosarcoma in a percutaneous biopsy tract has been reported. Therefore, it is important that the biopsy site be located so that it can be resected en bloc with the primary tumor. Because fine-needle aspiration has a diagnostic yield lower than core needle biopsy and does not obviate the need for sedation in the pediatric population, it is not a recommended approach for diagnosis.
Surgical or open biopsy of bone tumors can be incisional or excisional. Excisional biopsy is almost never performed in osteosarcoma because neoadjuvant chemotherapy can decrease tumor edema and, in doing so, simplify resection. In addition, time is required for adequate surgical planning, especially for younger children in whom placement of a growing prosthesis is being considered. Complications of open biopsy include anesthesia-related events, infection, bleeding, and tumor seeding in the biopsy tract. In addition, disruption of anatomic compartments and incorrect localization of the biopsy incision can lead to a more complex or more extensive tumor resection and can occasionally result in an amputation that would not have otherwise been necessary. As with percutaneous biopsy, the incision for an open biopsy should be placed and oriented in a way that allows it to be excised en bloc with the primary tumor. Complications of open biopsy, including those that result in more extensive resections, are unusual when the biopsy is performed at a center specializing in the diagnosis and treatment of malignant bone tumors.
In summary, the percutaneous and open biopsy routes are both acceptable. Given the implications for ultimate surgical resection of a poorly performed biopsy, biopsy should be performed or planned by an orthopedic surgeon with bone tumor expertise, ideally by the same orthopedic surgeon who will perform the ultimate resection. Percutaneous biopsies should only be performed by experienced interventional radiology staff working in conjunction with an experienced orthopedic surgeon. If the percutaneous route is recommended, it is important to counsel patients and families about the possible risks of core needle biopsy, including the potential for an inconclusive specimen. For a further discussion of the debate regarding the correct route for bone tumor biopsy, see Box 62-2 .
There are two possible approaches to obtaining a diagnostic specimen in suspected osteosarcoma—open biopsy and core needle biopsy. Considerable controversy exists regarding which diagnostic approach is best. An open biopsy is performed in the operating room by an orthopedic surgeon. As discussed in more detail in the text, an open biopsy should be performed by orthopedic surgeon with experience treating bone tumors and ideally by the same orthopedic surgeon who will ultimately perform the tumor resection. In a core needle biopsy, while imaging the tumor with CT, an interventional radiologist obtains several needle cores of tumor material. As with an open biopsy, a core needle biopsy should be performed by an interventional radiologist with expertise in the diagnostic evaluation of bone tumors. Planning for core needle biopsy should include a discussion with the orthopedic surgeon who will ultimately perform the resection. The relative merits of core needle and open biopsy as they relate to several core issues are discussed below:
1
Definitive Diagnosis
Because orthopedic surgeons directly visualize a tumor during an open biopsy and because pathologists often view frozen sections during open biopsies, nondiagnostic open biopsies are uncommon. In retrospective series, core needle biopsy results in a definitive, correct diagnosis in 78% to 94% of cases. The more recently published series report a diagnostic accuracy of 90% or more. The interventional radiology-guided core needle biopsies in these studies were performed in centers with considerable experience with the diagnosis of bone tumors and similar diagnostic accuracy would not be expected in inexperienced centers. Less tumor material is obtained from core needle biopsies, which can compromise the pathologist’s ability to perform additional testing, such as cytogenetics, in some cases.
2
Postprocedure Recovery
Both core needle and open biopsies require general anesthesia in children. In general, open biopsies are longer procedures, requiring a greater length of general anesthesia. Open biopsies are also more invasive and result in a longer postprocedure recovery.
3
Impact on Ultimate Resection
Whether the biopsy approach is core needle or open, definitive surgical treatment of osteosarcoma, which is essential for cure, requires excision of the biopsy tract or site en bloc with the primary tumor. A poorly placed biopsy can convert a tumor amenable to limb-sparing surgery into one that requires amputation. This impact on ultimate resectability is one of the greatest potential negative aspects of core needle biopsy. Because interventional radiologists are less knowledgeable about surgical approaches for definitive tumor resection in osteosarcoma, the risk of a poorly placed biopsy is potentially higher with a core needle biopsy. However, supporters of core needle biopsy argue that this risk can be reduced through discussion of the biopsy with the patient’s treating orthopedic surgeon.
4
Impact on Biologic Studies of Osteosarcoma
Although the availability of tumor material for the study of osteosarcoma biology is not an important issue from the perspective of the individual patient with osteosarcoma, it is an important issue from the perspective of researchers and future patients who could benefit from research discoveries. Because tumor resection occurs after the administration of chemotherapy, osteosarcoma resection specimens are often not suitable for laboratory investigation. After open biopsy there is usually sufficient prechemotherapy tumor material available for biologic studies after the diagnosis is made. This is not the case with core needle biopsies. Thus, progress in osteosarcoma research would likely be negatively affected if the majority of centers were to perform core needle biopsies.
Laboratory Evaluation
Laboratory test results are often normal in osteosarcoma. When laboratory abnormalities are present, they are nonspecific, having many other potential causes. The serum alkaline phosphatase level is elevated in approximately 50% of patients presenting with osteosarcoma. A higher percentage of patients with metastatic disease have a high alkaline phosphatase level, but this is not a reliable test of metastatic disease, because some patients with metastatic disease will have a normal result. As will be discussed later, alkaline phosphatase elevations are associated with a poorer prognosis. Lactate dehydrogenase (LDH) is elevated in approximately 30% of osteosarcoma patients. The erythrocyte sedimentation rate is generally not elevated in patients with osteosarcoma, whereas it is often elevated in those with Ewing sarcoma.
Pathology and Staging
Pathology
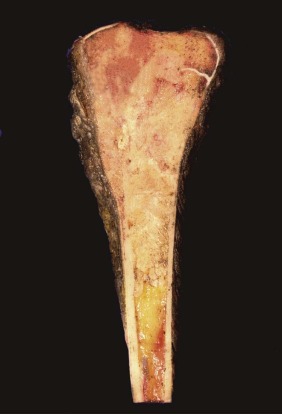
On gross examination, osteosarcoma usually presents as a large mass located in the metaphysis ( Fig. 62-9 ) arising in the medullary cavity and crossing the bony cortex, with invasion into the adjacent soft tissues. Microscopically, the malignant cells are pleomorphic but spindle cells typically predominate. Epithelioid, small round cells, multinucleated giant cells, and plasmacytoid cells may also be present and occasionally are the predominant morphology. Osteoid, which can be plentiful or sparse, is currently essential for the diagnosis of osteosarcoma. Sometimes, it is not present in the initial biopsy sample but is seen in the definitive resection specimen. If not present in either, the diagnosis of osteosarcoma cannot be made with certainty. The histologic appearance of osteoid is dense, pink, and amorphous. Classic or conventional osteosarcoma comprises 70% to 80% of cases of osteosarcoma. A number of other types of osteosarcoma are distinguished from classic osteosarcoma based on clinical or pathologic features ( Table 62-1 ).
| Type | Pathology | Approximate % (of all Osteosarcoma) |
|---|---|---|
| Classic | Anaplastic, numerous mitoses | 70-80 |
| Osteoblastic | Predominant matrix osteoid | 35 |
| Chondroblastic | Predominant matrix chondroid | 18 |
| Fibroblastic | Spindle cells in herringbone pattern | 18 |
| Jaw | Similar to classic osteosarcoma | 3-7 |
| Secondary | Similar to classic osteosarcoma | 7 |
| Telangiectatic | Cysts and septa, scant osteoid, giant cells present | 4-10 |
| Small cell | Small round malignant cells, osteoid present but quantity variable | 1-4 |
| Low-grade intramedullary | Minimal atypia, few mitoses | 1-2 |
| Parosteal | Located on bone surface, low grade, matrix osteoid and, in 50%, cartilage | 4 |
| Periosteal | Located on bone surface, intermediate grade, predominant matrix cartilage | 1-2 |
Classic osteosarcoma has a highly malignant appearance, with prominent anaplasia and frequent mitoses. There is variability in the histologic appearance of conventional osteosarcoma. To facilitate pathologic diagnosis and best describe this variability in appearance, classic osteosarcoma is divided into three subtypes based on the predominant matrix produced by the malignant cells. In the first, 50% of typical osteosarcoma is osteoblastic osteosarcoma, in which osteoid is the predominant matrix ( Fig. 62-10 ). Next, 25% of typical osteosarcoma is chondroblastic osteosarcoma in which cartilaginous islands are the predominant matrix. This subtype is differentiated from chondrosarcoma by the presence of osteoid. Because osteoid can be sparse in chondroblastic osteosarcoma, distinguishing chondroblastic osteosarcoma from chondrosarcoma can require extensive biopsy material, which may necessitate rebiopsy. Sixty-one percent of chondrosarcomas have mutations in the isocitrate dehydrogenase 1 and 2 genes ( IDH1 and IDH2 ). Mutations in these genes have not been seen in osteosarcoma. Therefore, the identification of an IDH1 or IDH2 mutation in a malignant bone tumor with a chondroid matrix would strongly suggest a diagnosis of chondrosarcoma. The remaining 25% of typical osteosarcoma is fibroblastic. In fibroblastic osteosarcoma, osteoid is minimal and spindle cells grow in a herringbone pattern. Many osteosarcomas have a mixture of different histologies, but in mixed histology cases one histology usually predominates. Of note, the subtypes of typical osteosarcoma were originally designated for the purpose of diagnosis, and their prognostic significance is uncertain. A better histologic response to chemotherapy in fibroblastic osteosarcoma and a poorer histologic response to chemotherapy in chondroblastic osteosarcoma have been consistent findings in several studies. Telangiectatic osteosarcoma also has a better histologic response to chemotherapy (see later). Whether these differences in histologic response to therapy are correlated with differences in OS is not entirely clear. In the largest study to evaluate this question, there was a slight, statistically significant 5-year OS advantage in patients with fibroblastic osteosarcoma. Unlike other types of osteosarcoma, histologic response to chemotherapy may not be correlated with outcome in chondroblastic osteosarcoma.
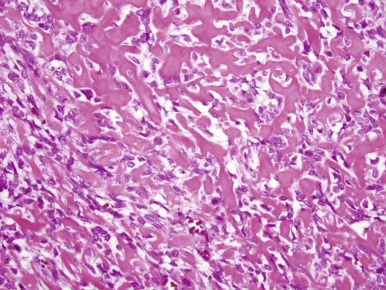
The remaining 20% to 30% of osteosarcomas that are not classic osteosarcomas are divided into secondary (occurring in Paget disease or after irradiation), jaw, telangiectatic, small cell, parosteal, periosteal, and low-grade intramedullary osteosarcoma. Telangiectatic osteosarcoma accounts for 4% to 11% of all osteosarcoma cases. The age and gender distribution are the same as in conventional osteosarcoma. Pathologic fracture is more common, occurring in 15% to 25% of cases. Radiographically, a lytic pattern is present because of the presence of large or small cystic areas and minimal osteoid formation. Microscopically, cysts are divided by septa that are lined with malignant cells producing scant osteoid. Cysts are also lined by benign, multinucleated giant cells. The presence of cysts and multinucleated giant cells can lead to misdiagnosis as an aneurysmal bone cyst or giant cell tumor, both benign bone tumors. Although case series published prior to the modern era of multiagent chemotherapy had reported that telangiectatic osteosarcoma has an adverse prognostic implication, more recent studies have reported a better histologic response to therapy and improved survival as compared with typical osteosarcoma.
Small cell osteosarcoma is a rare but histologically distinct variant representing 1% to 4% of osteosarcomas. The clinical and epidemiologic characteristics are the same as conventional osteosarcoma. Microscopically, tumors are composed of small round cells that produce variable amounts of osteoid. Differentiation from Ewing sarcoma and lymphoma can be difficult.
Osteosarcomas of the jaw, located in the maxilla or mandible, deserve specific mention. Even when secondary osteosarcomas are excluded, the mean age of patients with osteosarcoma in this location is the mid-30s, which is older than in classic osteosarcoma. These tumors are more often low grade; approximately 50% are low grade in most series. Histologically, the appearance is similar to that of classic osteosarcoma but a larger proportion of tumors are chondroblastic. In concordance with the greater proportion of low-grade tumors, prognosis is better for osteosarcoma of the jaw. Although local recurrence does occur, especially when the initial resection is inadequate, metastasis is unusual.
Low-grade intramedullary osteosarcoma is relatively rare, making up 1% to 2% of all osteosarcomas. Patients developing this form of osteosarcoma tend to be older than those with conventional osteosarcoma, with 30 years being the mean age at diagnosis. Microscopic examination reveals spindle cells permeating bony trabeculae, with minimal atypia and few mitoses. A variable amount of osteoid is present. Recurrence and metastasis are rare. With adequate resection, chemotherapy is generally not indicated.
Parosteal osteosarcoma is another low-grade osteosarcoma distinguished from low-grade intramedullary osteosarcoma by its location on the cortical surface of the bone. It is generally a sclerotic tumor attached to the bone by a broad base. Microscopically, the stroma is sparsely populated by minimally atypical spindle cells. The matrix contains osteoid and, in 50% of cases, cartilage. A limited degree of medullary involvement is seen in 25% of cases. Areas of dedifferentiation can be present at diagnosis or at the time of recurrence and portend a poorer prognosis. Of patients with dedifferentiation, 31% will die of disease, usually with pulmonary metastasis. Parosteal osteosarcoma makes up approximately 4% of all osteosarcomas. Like intramedullary low-grade osteosarcoma, parosteal osteosarcoma occurs in slightly older patients than conventional osteosarcoma and recurrence and metastasis are rare in adequately resected cases without dedifferentiation.
Parosteal osteosarcoma must be distinguished from periosteal osteosarcoma. Periosteal osteosarcoma also arises from the surface of the bone but is an intermediate- or high-grade lesion. Periosteal osteosarcoma has an age distribution similar to that of typical osteosarcoma but almost all tumors arise in the proximal tibia. The appearance on plain radiography is highly characteristic and suggests the diagnosis. Histology is of a spindle cell neoplasm with osteoid and cartilaginous islands present, usually in large amounts. The prognosis is better than in conventional osteosarcoma but probably not as good as in parosteal osteosarcoma without dedifferentiation or intermedullary low-grade osteosarcoma. There is controversy regarding the need for chemotherapy for periosteal osteosarcoma. The entity is too rare to permit conduct of prospective randomized controlled trials. Interpretation of retrospective series is difficult because variability in chemotherapy administration likely correlates with variability in clinicopathologic features related to outcome.
Secondary osteosarcoma arises in previously irradiated bone or in patients with Paget disease. In both cases, patients are older than those with conventional osteosarcoma. Pathology is similar to conventional osteosarcoma. Prognosis is poorer in Paget disease than in conventional osteosarcoma. There is some evidence that radiation-induced osteosarcomas have similar outcomes to de novo osteosarcoma if standard osteosarcoma chemotherapy is given.
Staging
Staging of osteosarcoma follows the Enneking or musculoskeletal society staging system ( Table 62-2 ). The TNM system is not used for several reasons. The TNM system lacks biologic relevance in that osteosarcoma seldom metastasizes to lymph nodes. In addition, it is more complex than needed for predicting outcome, because patients in different TNM stages have overlapping prognoses.
| Stage | Grade | Site | Metastasis | % of Patients | Surgical Margin | Chemotherapy | Representative Subtype |
|---|---|---|---|---|---|---|---|
| IA | G1 | T1 | M0 | 5 | Wide | No | Parosteal |
| IB | G1 | T2 | M0 | 3 | Wide | No | |
| IIA | G2 | T1 | M0 | 5 | Wide | Yes | Periosteal |
| IIB | G2 | T2 | M0 | 74 | Wide | Yes | Classic |
| III | G1, G2 | T1, T2 | M1 | 13 | Variable | Yes | Classic |
The Enneking staging system is based on tumor grade (G), site (T), and metastases (M). Low-grade (G1) tumors are well differentiated and have few mitoses, corresponding to Broders grades I and II. Osteosarcoma is high grade (G2) if it is poorly differentiated and has numerous mitoses, corresponding to Broders grades III and IV. As noted, most osteosarcomas are high grade (G2). Surgical site (T) is determined by whether the tumor is contained within its anatomic compartment of origin (T1) or extends beyond its compartment of origin (T2). For example, a typical osteosarcoma that arises in the medullary space and extends through the cortex into the soft tissue is T2. A parosteal osteosarcoma that does not extend from the bone surface, its site of origin, into the medullary space and does not invade the surrounding soft tissue is T1. Most classic osteosarcomas are T2. Metastases are absent (M1) or present (M2). Stages IA and IB are low-grade (G1) tumors. Stages IIA and IIB are high-grade tumors. Stage IA tumors are low-grade osteosarcomas that are T1 or lack extension beyond the compartment of origin. Stage IB tumors are low-grade osteosarcomas that are T2 or demonstrate extension beyond the compartment of origin. Similarly, stage IIA tumors do not have extracompartmental extension, whereas IIB tumors do. Stage III osteosarcomas are tumors of any grade and any site that have distant metastases. The vast majority of osteosarcomas are stage IIB. The next most common stage for osteosarcoma at presentation is stage III. Stages IA, IB, and IIA are uncommon, with 3% to 5% of osteosarcomas falling into each of these categories.
Natural History and Prognosis
The most significant predictor of survival in osteosarcoma is the presence of residual disease caused by incomplete resection of primary tumor in localized osteosarcoma, local recurrence, or the presence of metastases. Consequently, the natural history, prognosis, and prognostic factors of localized, relapsed, and metastatic osteosarcoma differ significantly.
Localized Disease
Natural History
Prior to the use of chemotherapy for osteosarcoma, the OS of patients with localized disease who had a complete resection was 20%. Death was almost always caused by complications of pulmonary metastases. With wide or radical surgical margins, local recurrence was rare. The development of pulmonary metastases in 80% of patients treated with only complete resection of the primary tumor has been interpreted as evidence of the presence of micrometastatic foci in the lungs at the time of diagnosis and was one rationale for adjuvant chemotherapy.
Five-year event-free survival (EFS) and OS of localized osteosarcoma in the era of modern therapy are 65% to 75% and 70% to 80%, respectively. Approximately 80% of relapses are pulmonary either alone or, in 25% of patients, with a second disease site, which is most often bone. Bone, distant from the initial resection, is the initial relapse site in 8% of patients; 1% to 2% of patients have recurrence isolated to unusual sites such as brain, kidneys, heart, or liver. Local recurrence is seen in 2% to 8% of patients, despite having had adequate surgical margins. Most relapses occur during the first 2 years after completion of chemotherapy. However, relapses occur as late as 10 years and the EFS decreases slowly between 2 and 10 years. In the few studies reporting long-term follow-up data, EFS decreases to 60% at 10 years from 65% at 5 years.
Prognostic Factors
The most consistent predictor of outcome in localized, fully resected osteosarcoma is the extent of tumor necrosis after neoadjuvant chemotherapy. When investigators at the Memorial Sloan-Kettering Cancer Center first gave neoadjuvant, or preoperative, chemotherapy to facilitate surgical resection without amputation, they noted varying degrees of necrosis on pathologic examination of resected tumor specimens. It was later observed that patients with significant necrosis had a better EFS and OS than those with lesser degrees of necrosis. However, tumor response is not a true prognostic factor because it cannot be determined until after the initiation of chemotherapy.
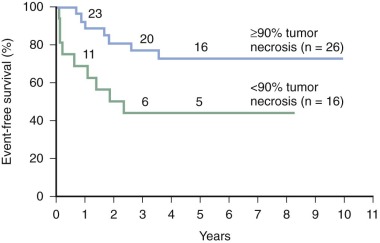
The generally accepted grading system for tumor necrosis ( Table 62-3 ) assigns higher grades to tumors with greater evidence of necrosis. In most studies, grades III and IV, in which more than 90% of the tumor is necrotic, define the group of patients whose tumors have a good response to chemotherapy. Patients whose tumors have a poor response have 10% or more viable tumor, corresponding to grades I and II. Patients with a poor response have disease-free survival (DFS) rates of 40% to 50%, whereas those with a good response have DFS rates of 70% to 80% ( Fig. 62-11 ). An improved outcome for poor responders is an objective of past and current trials (see “ Chemotherapy for Localized Disease ”).
| Response | Grade | Histology |
|---|---|---|
| Poor | I | Necrosis minimal or absent |
| II | Necrosis is <90% of the tumor but greater than minimal | |
| Good | III | Scattered areas of viable tumor but >90% of tumor necrotic |
| IV | No viable tumor |
Radiographic parameters have been investigated for correlation with tumor necrosis and outcome. A decrease in FDG-PET standard uptake value (SUV) before and after neoadjuvant chemotherapy is correlated with histologic response. Neither the change in SUV nor absolute SUV after neoadjuvant chemotherapy has a statistically significant association with OS in localized osteosarcoma. The lack of statistical significance may be due to investigation in only a small population of patients, and larger studies are needed to determine whether FDG-PET responses are predictive of outcome in osteosarcoma. Various MRI techniques including dynamic contrast-enhanced MRI and diffusion-weighted MRI show promise for potential to predict histologic response.
Additional prognostic factors have been identified in localized osteosarcoma. In many cases these factors are not consistently demonstrated to be statistically significant across studies. This may be due to a small effect size and, as a result, the need for large studies to identify a factor as being statistically related to outcome. In other cases, differences in measurement or categorization contribute to variable results across studies.
Tumor size has been identified as a significant prognostic factor by multivariate analysis in several studies. However, other studies have not found this patient variable to be predictive of outcome. One possible explanation for this conflict is variability in the definition of tumor size and measurement techniques. Some studies evaluate absolute tumor length, others use relative (to the entire bone) tumor length, and still others base analyses on tumor volume. Most studies showing size to be a significant predictor of outcome on multivariate analysis have measured tumor volume. However, the volume that distinguishes small, low-risk tumors from large, high-risk tumors varies among studies. In summary, further studies using standard measurement techniques and risk criteria need to be conducted before tumor size can be used as a clinically significant predictor on which to counsel patients or base therapeutic decisions. The prognostic significance of tumor size might be caused by the influence of tumor size on resectability. In other words, larger tumors may confer a poorer prognosis because it is less likely that the tumor will be completely resected with wide margins.
Several studies have found an association between tumor site and outcome, with axial and proximal appendicular primary tumors having a worse prognosis than distal extremity tumors. However, there are potential interactions between tumor site and other osteosarcoma prognostic factors. In particular, site and size are likely to be associated. A distal extremity osteosarcoma is likely to be noticed at a smaller size because of the relative lack of soft tissue. More importantly, the higher risk sites, such as axial skeleton, are also those for which a complete resection is less likely.
Elevated LDH and alkaline phosphatase levels at the time of diagnosis are associated with a poorer prognosis. In one study, the EFS of patients with a normal alkaline phosphatase level at diagnosis was 88%, whereas it was 46% for patients with an elevated alkaline phosphatase level. There are several issues that limit the usefulness of LDH and alkaline phosphatase as prognostic indicators. First, elevated levels are not consistently associated with a poorer prognosis in all studies. Second, elevations in LDH and alkaline phosphatase levels confer only a modestly increased relative risk (1.5 for LDH and 2 for alkaline phosphatase) of a poor outcome.
Other prognostic factors that have been reported to be significant in a limited number of studies are age and gender, with older age and male gender being associated with a worse outcome. Chemotherapy toxicity, represented by grade 3 or 4 mucositis, has been found to be correlated with improved OS. A single study from the Children’s Oncology Group (COG) identified a high body mass index at the time of diagnosis as a risk factor for inferior survival.
As noted earlier, histologic subtype may be a prognostic factor. P-glycoprotein positivity, loss of heterozygosity at the RB1 locus, and high ezrin expression have been identified as possible biology-based predictors of a poor outcome. These prognostic factors are discussed more extensively (see “ Biology ”).
In summary, the extent of tumor necrosis is the most reliable prognostic indicator in localized, completely resected osteosarcoma. Additional factors correlated with a poorer outcome include older age, less chemotherapy toxicity, larger tumor size, axial tumor site, elevated LDH and alkaline phosphatase levels, and high body mass index. However, for prognostic factors other than tumor necrosis to be utilized for risk stratification, further validation is required in large populations who received uniform therapy. Identification of reliable prognostic factors available at the time of diagnosis is a research priority in osteosarcoma. Biologic efforts such as TARGET (see “ Genomics ” under “ Biology ”) and further investigation of imaging characteristics are most likely to provide robust predictors of outcome.
Metastatic and Relapsed Disease
The OS for patients with relapsed disease is poor, with most series showing a 25% OS at 5 years. As with primary osteosarcoma, the most significant prognostic factor for postrelapse survival is the ability to achieve complete resection of all disease sites. In one report of 162 patients with initially localized osteosarcoma who developed recurrent disease, the 5-year projected OS was 39% for those who had complete resection of all disease and 0% for those who did not. With isolated pulmonary metastases, prognosis is significantly influenced by the number of pulmonary nodules and whether there is unilateral or bilateral pulmonary disease. In particular, having more than one pulmonary nodule at relapse conveys a worse prognosis. An additional prognostic factor is the duration of remission. Patients experiencing relapse less than 24 months after initial diagnosis have a worse outcome. Finally, use of chemotherapy is associated with a slightly improved postrelapse prognosis, particularly in patients with unresectable disease. Patients who experience second and subsequent recurrences have a 5-year OS rate of about 15%. As with the initial recurrence, the duration of relapse-free interval, the number of lesions at recurrence, and the ability to resect recurrent disease are all associated with outcome.
Local recurrence warrants special mention because of its dismal prognosis. The 5-year DFS is 0% to 15%. Local recurrence is almost always associated with concurrent or delayed pulmonary metastasis. In one series, the outcome of patients with local recurrence was strongly influenced by the presence or absence of systemic metastases at the time of local recurrence. The 5-year DFS was 25% for patients without systemic metastases at the time of local recurrence, and it was 0% for those with systemic metastases.
The outcome and prognostic factors for metastatic disease present at diagnosis are similar to those for relapsed disease. The 5-year OS of patients presenting with primary metastatic disease is 20% to 30%. One exception to the poor prognosis for patients with metastatic disease is skip metastases. In patients with skip metastases, OS is 50%, even when pulmonary metastases are also present. Higher OS has been reported in some chemotherapy trials, possibly because of therapy efficacy or shorter periods of follow-up. An important predictor of a poor outcome is the inability to achieve complete surgical remission. Similarly, patients with metastases to more than one site or more than one organ are at increased risk for death from disease. Lymph node metastasis at the time of diagnosis is uncommon, occurring in less than 5% of patients. Lymph node involvement at the time of diagnosis is associated with larger tumor size and an extraskeletal primary lesion. OS at 5 years for those with lymph node metastasis at diagnosis is 10%. In one study, an alkaline phosphatase level higher than 500 IU/L was significantly associated with poor prognosis on multivariate analysis.
Biology
Structural chromosomal alterations, loss of tumor suppressor function, and oncogene amplification have all been described in osteosarcoma. In addition, alterations in specific proteins contribute to chemotherapy resistance and metastatic behavior. A summary of genes altered in osteosarcoma is presented in Table 62-4 .
| Gene | Alteration | Gene Function | % of Sporadic Osteosarcoma |
|---|---|---|---|
| RB1 | Deletion, inactivation | Cell cycle control | 70 |
| TP53 | Mutation, inactivation | Apoptosis Response to DNA damage Cell cycle control | 50 |
| MDM2 | Gene amplification | TP53 regulation | 10-15 |
| COPS3 | Gene amplification | TP53 regulation | 25 |
| CDKN2A | Deletion, promoter methylation | Cell cycle control through inhibition of CDK4-CCND1 | 15-20 |
| MET | Increased expression | Motility Invasion | 60 |
| FOS | Increased expression | Proliferation Possibly, differentiation | 60 |
| MYC | Gene amplification | Proliferation | 40 |
| ABCB1 | Increased expression | Efflux of chemotherapeutic agents | 25-45 |
| DHFR | Increased expression | Folate metabolism; MTX target | 60% relapsed tumors |
| ERBB2 | Increased expression | Growth factor receptor | 40 |
| Ezrin | Increased expression | Membrane cytoskeleton linker, facilitates metastasis | 92 |
| FAS | Decreased expression | Apoptosis | 60% pulmonary metastases lack FAS |
| Reduced folate carrier | Decreased expression | MTX transport | 65 |
| IGF1R | Increased expression | Growth factor receptor, proliferation | Unknown |
Cell of Origin
Genetically engineered murine models of osteosarcoma have helped define the cell of origin for osteosarcoma. Lineage-specific models have demonstrated that osteosarcoma can arise from mesenchymal stem cell progenitors, osteoblast progenitors, and osteoblasts. In these lineage-specific genetically engineered mouse models, osteosarcoma develops with the highest penetrance and shortest latency when derived from osteoblast progenitors whereas mesenchymal stem cell progenitors give rise to a larger proportion of poorly differentiated soft tissue sarcomas. A case report of osteosarcoma arising from donor cells after allogeneic stem cell transplantation suggests that mesenchymal stem cells can give rise to osteosarcoma in humans as well.
Genomics
Osteosarcomas contain complex genomic aberrations, suggesting the involvement of multiple genes in osteosarcoma development. Copy number abnormalities, changes in ploidy, and structural rearrangements are seen in all high-grade osteosarcomas. It is a tumor type in which chromothripsis has found to occur frequently. Homogeneously staining regions and marker, double-minute, and ring chromosomes are seen in most tumors. A representative karyotype and Circos plot are shown in Figure 62-12 . Nonclonal changes, often superimposed on clonal aberrations, are present in many tumor specimens. Several comprehensive next-generation sequencing projects are underway, including one being conducted as part of the National Cancer Institute sponsored Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project. The nonclonal complex nature of the osteosarcoma genome makes the interpretation of genomic data difficult and the identification of candidate genes implicated in a significant proportion of osteosarcomas challenging.
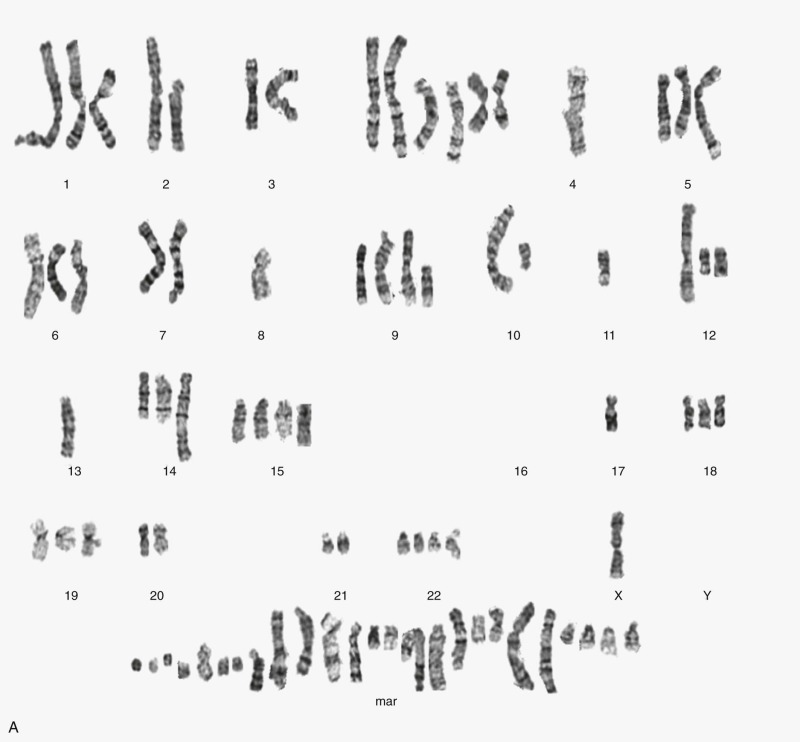
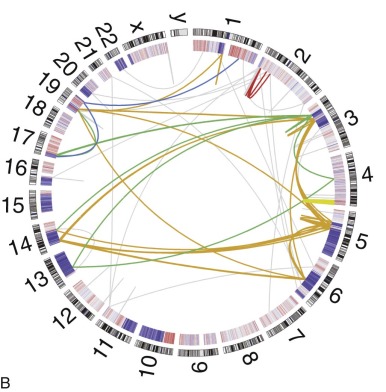
Osteosarcoma DNA ploidy ranges from diploid to hexaploid. Spectral karyotyping (SKY) of osteosarcomas reveals an average of 39 chromosomal rearrangements/tumor. Chromothripsis is the simultaneous occurrence of multiple somatic rearrangement events during one single cellular episode. In nine osteosarcoma samples, three (33%) had evidence of chromothripsis. Chromoplexy is a related mechanism of chromosomal rearrangement in which series of interrelated rearrangements affect multiple chromosomes. Preliminary results of an ongoing comprehensive next-generation sequencing study found evidence of chromoplexy in most osteosarcoma samples studied. Pilot studies have demonstrated the feasibility of using detection of patient-specific translocations to track disease burden in patients with osteosarcoma and other solid tumors. Whether this method for detection of minimal residual disease burden will have prognostic implications in osteosarcoma as it has in leukemia remains to be determined.
Multiple genomic techniques have been applied to the assessment of copy number alterations in osteosarcoma. Traditional and spectral karyotype permitted the initial identification of genomic complexity and copy number alterations in osteosarcoma. More sophisticated techniques such as array comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) arrays have permitted the identification of recurrently amplified and deleted regions. In almost all CGH analyses, gains are more common than losses. Amplified regions consistently seen in a significant proportion of tumors and across studies include 1q21-q24, 6p11.2-p12, 8q21.3-q24, 17p11.2-p12 (high-level amplification), 1p, 1q21, 5p14, and 6p12-21.3. Commonly seen deletions include 2q and 6q and chromosomes 9, 10, and 13q. Copy number alterations are also present in other regions but have been reported in a small proportion of cases or single studies only. Loss of 13q14 and gain of 8q (8q21.3-q22 or 8cen-q13) have been associated with a worse prognosis, and 5q loss has been associated with a better prognosis, although these findings have not been replicated. Higher-detail analysis with fluorescence in situ hybridization (FISH), quantitative polymerase chain reaction (PCR) assay, or array CGH has revealed that amplification and deletion patterns are complex, in that individual genes in an amplified region may not be amplified.
Importantly, candidate genes have been identified via assessment of copy number in osteosarcoma. The region 12q13-14, which is amplified in 10% of osteosarcomas, contains multiple potential oncogenes, including MDM2. In the commonly amplified region 17p11.2-p12, candidates PMP22, TOP3A, MAPK7, and COPS3 display high levels of amplification in more than 50% of cases using semiquantitative PCR assay and microsatellite markers. RB1 is located at 13q14, a region that is lost in many osteosarcomas. MDM2 and COPS3 amplifications and RB1 loss are discussed further later. Three groups have identified a recurrent focal deletion at 3q13.3 including the candidate tumor suppressor gene LSAMP. The MYC gene, located at 8q24.1, is amplified in as many as 40% of osteosarcomas. Amplification of 8q24 containing MYC and of 12q14 harboring CDK4 and a higher overall degree of loss of heterozygosity have been associated with a poorer outcome. As with other prognostic factors in osteosarcoma, the prognostic significance of these copy number alterations needs to be validated in larger, prospectively conducted clinical studies.
Investigation into the relationship between aberrations in microRNAs and osteosarcoma pathogenesis and prognosis are in their infancy. Definitive conclusions await research with large numbers of osteosarcoma samples and more robust statistical methodologies.
Tumor Suppressors
RB1
The retinoblastoma gene RB1 , located at 13q14, was the first tumor suppressor gene identified and is one of the most frequently altered genes in osteosarcoma. As noted earlier (see “ Etiology ”), the second most common tumor in patients with hereditary retinoblastoma who carry germline RB1 mutations is osteosarcoma. RB1 abnormalities are also present in most sporadic osteosarcomas. RB1 inactivation is believed to contribute to osteosarcoma tumorigenesis through its role in cell cycle control and possibly through its regulation of differentiation and apoptosis.
Hereditary retinoblastoma is a condition in which individuals with a heterozygous germline RB1 mutation develop retinoblastoma, a malignant tumor of the embryonal neural retina, with 90% penetrance. Germline RB1 mutations can be sporadic or inherited. RB1 germline mutations in patients who develop bilateral tumors are usually small deletions or frameshift mutations resulting in a null protein, whereas 40% of RB1 germline mutations in patients with unilateral tumors are in-frame point mutations. The cumulative incidence of a second malignancy by 50 years after the initial retinoblastoma diagnosis is 51%. Approximately 50% of the second malignancies developing in patients with hereditary retinoblastoma are osteosarcomas. Both osteosarcomas and retinoblastomas occurring in patients with hereditary retinoblastoma have somatic loss of the normal RB1 allele, with resulting complete absence of a functional RB1 protein.
Somatic alterations in the RB1 gene are seen in most sporadic osteosarcomas. Loss of heterozygosity at the RB1 locus is seen in 65% of sporadic osteosarcomas. Structural changes, usually associated with loss of heterogeneity, are present in 30% to 40% of tumors, and homozygous deletions are seen in 23% of tumors. Point mutations are rare and are present in only 6% of cases. Overall, 70% of sporadic osteosarcomas have at least one RB1 abnormality, and many tumors have a combination of RB1 alterations. RB1 gene alterations are generally not seen in low-grade bone tumors. There is some evidence that the initially mutated allele is usually of paternal origin and the allele that undergoes deletion is maternally derived. Decreased RB1 expression is seen in about 50% of osteosarcomas, but changes in RB1 expression are not correlated with RB1 gene alterations. For example, in some tumors with RB1 loss of heterogeneity, RB1 protein levels are not reduced.
Some studies have found an association between RB1 loss of heterogeneity and a poorer prognosis. However, these studies were based on univariate analyses in relatively small populations and another study had contradictory results. Theoretically, RB1 inactivation could contribute to a poorer prognosis through increasing the expression of thymidylate synthase and dihydrofolate reductase, consequently increasing resistance to antimetabolites, an important component of osteosarcoma therapy.
As reviewed by Classon and Harlow, RB1 is a key regulator of the G1 to S cell cycle transition. RB1, by binding and thereby inhibiting the E2F family of transcription factors, blocks the transition from the G1 to the S phase of the cell cycle. RB1 is regulated by cyclin-dependent kinases (CDKs), particularly CDK4 and CDK6, which interact with CCND1. These CDK-CCND1 complexes phosphorylate RB1. RB1 phosphorylation inhibits the RB1-E2F interaction, allowing transcriptional activation by E2F, which leads to progression from G1 to S ( Fig. 62-13 ). RB1 probably also controls cell cycle progression through chromatin remodeling, as reviewed by Khidr and Chen. In particular, the RB1-E2F complex, by interacting with histone deacetylases, histone methyltransferases, and DNA methylases, contributes to the inactivation of E2F target genes during G0 and G1.
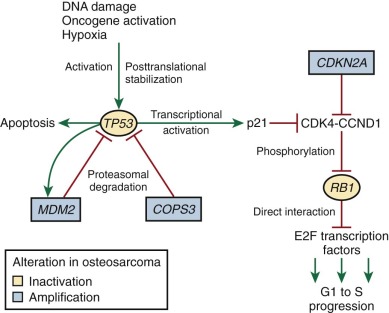
Genetically engineered Rb1 null mouse models implicate Rb1 in regulation of cellular differentiation and apoptosis. Rb1 deletion in mice results in embryonic lethality due to a placental defect. When rescued from the placental defect, abnormal bone development is noted, and this appears to be due to increased proliferation in osteoblast cells combined with a failure in terminal osteoblastic differentiation. Through interaction with CBFA1, a key transcriptional regulator in osteoblasts, RB1 induces osteogenic differentiation. Loss of RB1 in osteoblast precursors appears to result in dedifferentiation, as demonstrated by an increased propensity for adipogenic differentiation. The significance of RB1 differentiation and apoptosis roles in tumorigenesis in general and osteosarcomagenesis in particular is not clear.
TP53
There is convincing evidence that abnormal TP53 function is one of the central events in osteosarcomagenesis. Patients with Li-Fraumeni syndrome, who carry heterozygous germline TP53 mutations, are predisposed to osteosarcoma. Somatic alterations in the TP53 gene are found in a significant percentage of sporadic osteosarcomas. Furthermore, several different transgenic mouse models, with altered or absent TP53 function, develop osteosarcoma. Finally, components of the TP53 regulatory and effector pathways, MDM2 (murine double-minute 2), CDKN2A (cyclin-dependent kinase inhibitor 2A), and COPS3 (COP9 constitutive photomorphogenic homolog subunit 3) are also frequently perturbed in osteosarcoma, particularly in tumors lacking TP53 mutations.
Of cancers arising in patients with Li-Fraumeni syndrome, 10% are osteosarcoma, making osteosarcoma the second most common tumor in Li-Fraumeni syndrome. In addition to osteosarcoma, patients with Li-Fraumeni syndrome are predisposed to breast cancer, various soft tissue sarcomas, brain tumors, and various carcinomas. TP53 germline mutations are present in 7% of osteosarcoma patients who have a personal history of multiple cancers but who do not meet Li-Fraumeni diagnostic criteria. Individuals with Li-Fraumeni syndrome have germline mutations in TP53 located throughout the gene, although approximately 75% of the mutations are in exons 5 to 8, which encode the DNA-binding domain of the TP53 protein. Tumors arising in patients with Li-Fraumeni syndrome typically have loss of the normal TP53 allele.
Somatic alterations of the TP53 gene are present in 20% to 50% of sporadic osteosarcomas. Despite the prevalence of somatic TP53 mutations in sporadic osteosarcomas, germline TP53 mutations are rare in patients with sporadic osteosarcoma without a personal or family history of multiple cancers. The wide range in the reported frequency of somatic TP53 alterations in sporadic osteosarcoma is the result of small study size, differences in techniques of ascertainment, and varying definitions of an abnormal result. TP53 point mutations are present in 20% to 30% of osteosarcomas. As with TP53 mutations in other cancer types, missense mutations constitute approximately 75% of osteosarcoma point mutations. Point mutations are present throughout the TP53 coding sequence, although, as with Li-Fraumeni syndrome germline mutations, approximately 73% occur in exons 5 to 8. The initial TP53 abnormality to be recognized in osteosarcoma was gross rearrangements of the TP53 gene. Gross rearrangements are present in 15% to 25% of cases. Studies that evaluated TP53 for both point mutations and gross gene rearrangements have found that almost 50% of osteosarcomas examined have aberrant TP53 . Because mutant TP53 protein usually accumulates in cells because of an increased half-life, TP53 immunohistochemistry has been used to screen for TP53 mutations in osteosarcoma. By immunohistochemistry, 15% to 25% of osteosarcomas have TP53 abnormalities, reflecting the fact that immunohistochemistry has a relatively low sensitivity for detecting TP53 mutations. Approximately 50% of osteosarcomas with TP53 point mutations have loss of the normal allele.
Several genetically engineered mouse models with dysfunctional or absent Tp53 develop osteosarcoma. In a genetically engineered mouse model in which Tp53 and Rb1 deletion is restricted to osteoblasts, osteosarcoma develops with a high penetrance and short latency. Mice homozygous for null Tp53 develop a variety of tumors. Although lymphoma and hemangiosarcoma are the most common tumor types seen, 4% of the mice develop osteosarcoma. Mice heterozygous for null Tp53 develop tumors at 25% the rate and at an older age than homozygous null mice, but 25% of the tumors that develop are osteosarcomas. Osteosarcomas are much more common in mice genetically engineered to be heterozygous for a common cancer-associated Tp53 mutation, R172H. Almost 50% of these mice develop osteosarcoma, usually associated with hematogenous metastases. Several other mouse models heterozygous for mutant Tp53 develop osteosarcoma at a rate of 20% to 30%. Metastases are common in these models as well. Transgenic mice expressing the SV40 T antigen from different promoters develop osteosarcoma at a rate of approximately 70%. The mechanism of transformation in the SV40 T antigen–expressing models probably involves the inactivation of Tp53 and Rb1.
In other cancers, TP53 mutation status has been associated with proliferation, response to chemotherapy, and outcome. In osteosarcoma, the response to chemotherapy is equivalent in TP53 mutant and TP53 wild-type osteosarcomas. Several studies have examined the impact of TP53 mutation on survival in osteosarcoma. Only one study, in 30 patients, showed a worse survival in patients whose tumors had TP53 mutations. The remainder of the studies did not show a relationship between TP53 mutation status and outcome. Finally, TP53 mutations are not more common in metastatic disease than in localized disease. Actually, the TP53 mutation status of primary and metastatic tumors tends to be concordant.
As reviewed by Levine and Vogelstein and colleagues, TP53 mutations and structural alterations lead to transformation by interfering with TP53 tumor suppressor functions and, possibly, by producing a TP53 protein that has gained oncogenic function. TP53 tumor suppressor activities include cell cycle control and induction of apoptosis. Several forms of cellular stress, including DNA damage, hypoxia, shortened telomeres, and oncogene activation lead to TP53 posttranslational stabilization and TP53 activation. After activation, TP53 binds DNA and promotes the transcription of several genes, including CDKN1A and MDM2 . As in other tumor types, the TP53 mutations seen in sporadic osteosarcomas and Li-Fraumeni syndrome are concentrated in the DNA-binding domain and interfere with the transcriptional activity of the protein. TP53 prevents progression from the G1 phase to the S phase of the cell cycle through the actions of one of its target genes, CDKN1A . The CDKN1A protein inhibits the CDK4-CCND1 complex, which in turn keeps RB1 in an unphosphorylated state. Unphosphorylated RB1 binds E2F transcription factors, preventing progression into the S phase (see Fig. 62-13 ). TP53 also plays a regulatory role in the G2 to M cell cycle checkpoint. Another inhibitor of CDK4-CCND1 is CDKN2A. CDKN2A abnormalities are also seen in osteosarcoma (see later).
As with TP53-mediated cell cycle control, TP53-mediated apoptosis is triggered by DNA damage. In addition, oncogene overexpression and RB1 inactivation stimulate TP53-mediated apoptosis. The pathways involved in TP53-mediated apoptosis are not fully understood. However, they likely involve proapoptotic proteins whose genes are targets of TP53 transcriptional activation, including BAX, PMAIP1, and TP53AIP1.
TP53 inactivation allows cells to progress through the cell cycle and escape apoptosis, despite the presence of DNA damage. Consistent with this observation, the presence of a TP53 mutation in osteosarcoma is significantly correlated with a greater level of genomic instability. Thus promotion of genomic instability and the resultant accumulation of secondary mutations in oncogenes or tumor suppressors is yet another mechanism of transformation in TP53 mutant tumors.
TP53 Regulators: MDM2 and COPS3
The regulators of TP53 activity, MDM2 and COPS3, are amplified in a proportion of osteosarcomas. MDM2 inhibits TP53 activity by direct interaction with the TP53 DNA-binding domain and by transporting TP53 to the cytoplasm. In addition, MDM2 ubiquitinates TP53, targeting it for proteasomal degradation. MDM2-mediated ubiquitination is enhanced by DNA damage. The MDM2 gene, located at 12q14, is amplified in 10% to 15% of osteosarcomas. Usually, MDM2 amplification occurs in tumors without TP53 mutations, and thus MDM2 amplification acts as an alternative mechanism of TP53 inactivation. There is limited evidence that MDM2 amplification is more common in parosteal and other forms of low-grade osteosarcoma.
COPS3, located at 17p11.2, a frequently amplified region in osteosarcoma, is a subunit of the COP9 signalosome. The COP9 signalosome interacts directly with TP53 and, through phosphorylation, targets TP53 for interaction with MDM2 and degradation by the 26S proteasome. COPS3 gene amplification is present in 25% to 30% of osteosarcomas. Increased COPS3 expression through alternative mechanisms is present in some tumors lacking COPS3 gene amplification. Like MDM2 amplification, COPS3 overexpression may be an alternative mechanism of TP53 inactivation. TP53 mutations are present in a small proportion of COPS3- amplified osteosarcomas.
CDKN2A and CDKN2D
As described earlier, CDKN2A, like the TP53-inducible protein CDKN1A, is an inhibitor of the CDK4-CCND1 complex. As with CDKN1A, inhibition of CDK4-CCND1 by CDKN2A prevents progression from the G1 to the S phase. Ten to 20 percent of osteosarcomas have CDKN2A homozygous deletion, and in another 5% to 10% the CDKN2A promoter is methylated. Similarly, by immunohistochemistry, 16% of osteosarcomas lack CDKN2A protein expression. CDKN2A deletions appear to be more common in osteosarcomas grown in cell culture but seem to be less common in osteosarcomas with RB1 abnormalities, possibly because the two alterations have a similar effect on cell cycle control. Few studies have evaluated the coexistence of TP53 mutations and CDKN2A deletions in osteosarcomas. There is limited evidence that the two abnormalities occur simultaneously.
CDKN2D is an alternatively spliced product of the gene encoding CDKN2A. CDKN2D inhibits MDM2 by direct binding. Thus loss of CDKN2D leads to increased MDM2-mediated TP53 degradation. As a consequence of the gene structure, the 10% to 20% of osteosarcomas with CDKN2A homozygous deletion also lack functional CDKN2D.
RECQ DNA Helicases
As noted earlier, osteosarcoma incidence is increased in three autosomal recessive cancer predisposition syndromes caused by RECQ DNA helicase mutations, Rothmund-Thomson syndrome, Werner syndrome, and Bloom syndrome. The overlapping but distinct clinical features and specific nature of osteosarcoma predisposition in these syndromes has already been discussed (see “ Etiology ”). Whereas germline RECQL4 mutations predispose patients with these syndromes to osteosarcoma, somatic RECQL4 mutations are not present in sporadic osteosarcomas.
Patients with Rothmund-Thomson syndrome caused by mutations in RECQL4 develop osteosarcoma with a prevalence of approximately 30% and probably do not have an increased susceptibility to other types of cancer. Interestingly, osteosarcoma occurs only in the 60% of patients with Rothmund-Thomson syndrome who have truncating or inactivating RECQL4 mutations. On the other hand, patients with Bloom and Werner syndromes, caused by mutations in RECQL2 and RECQL3, respectively, are prone to a wide variety of cancers, and osteosarcoma represents less than 10% of the cancers occurring in these patients. Why RECQL4 mutations, in contrast to RECQL2 and RECQL3 mutations, specifically increase the risk for osteosarcoma is unknown.
The mechanism whereby loss of function of RECQ DNA helicases results in tumor development generally and in osteosarcoma in particular are not well understood. Proposed RECQ functions that could be related to tumor formation include participation in DNA recombination, maintenance of chromosome stability, recovery from DNA replication collapse, initiation of DNA replication, and telomere processing. The three RECQ DNA helicases mutated in cancer predisposition syndromes have overlapping but not identical functions. In vitro cell culture and mouse models of RECQL4 mutant Rothmund-Thomson syndrome display increased chromosomal instability with high rates of aneuploidy. Bloom and Werner syndrome models have abnormally high rates of homologous recombination events, with accumulation of recombination intermediates. Studies with the Xenopus RECQL4 homologue have suggested that RECQL4 participates in the initiation of DNA replication. Several lines of evidence have indicated that the RECQ helicases likely play an essential role in reestablishing DNA replication after disruption of the replication fork. Finally, WRN may be involved in telomerase-independent telomere maintenance.
WWOX
WWOX encodes a WW domain containing oxoreductase. This class of proteins has a wide variety of functions including regulation of protein degradation, transcription, and splicing. There is evidence from other cancers that WWOX can act as a tumor suppressor. Mice carrying a homozygous deletion of Wwox develop osteosarcoma at a rate of 30% prior to premature death at 4 weeks postnatal age. Thirty percent of human osteosarcoma samples have WWOX deletions, and 60% have reduced or absent WWOX expression. WWOX interacts directly with RUNX2, a regulator of osteoblast differentiation. In osteosarcoma cell lines deficient in WWOX expression, forced WWOX expression results in decreased RUNX2 expression, decreased proliferation, decreased invasion, and a reduction in xenograft viability.
Developmental Pathways and Stem Cell Programs
Wnt Pathway
The Wnt pathway is active in bone development. Activating mutations in LRP5 (low density lipoprotein receptor–related protein 5), which encodes an Wnt coreceptor (with frizzled4), cause high bone mass whereas inactivating LRP5 mutations result in decreased bone mass. Despite multiple investigations into Wnt pathway activity in osteosarcoma, it remains unclear how and whether perturbations in Wnt pathway activity contribute to osteosarcomagenesis. The majority studies conclude that overactive Wnt signaling contributes to osteosarcoma formation. For example, deletion of the Wnt inhibitory factor, Wif1, in mice increased osteosarcoma formation after irradiation. However, there are conflicting reports such as a genome-wide expression study finding downregulation of the Wnt pathway in osteosarcoma.
USP1
USP1 encodes a deubiquitinating enzyme that deubiquitinates inhibitor of DNA-binding/differentiation (ID) proteins, preventing their degradation. ID proteins or inhibitors of DNA binding are basic helix-loop-helix transcription factors that have been shown to inhibit differentiation and maintain a stemlike state. In a small group of osteosarcoma tumors, expression of USP1 and ID2 were correlated and 7 of 14 tumors had high USP1 and ID2 expression. In U2OS and 143B osteosarcoma cell lines USP1 knockdown decreased ID1, ID2, and ID3 protein levels; decreased expression of the stem cell markers e-cadherin and fibronectin; and increased expression of markers of mature osteoblasts. USP1 knockdown decreased the number of cells in S phase of the cell cycle and decreased xenograft formation. If the results of this single study are validated, USP1 inhibition is a potential therapeutic avenue for osteosarcoma. Development of USP1 inhibitors is of interest owing to potential for activity in a wide variety of cancers, including lung cancer.
Oncogenes
Multiple cell surface receptor oncogenes are expressed and activated in osteosarcoma. It appears as though coexpression and coactivation of multiple cell surface receptors is a feature of osteosarcoma cell lines. Whether this phenomenon is also present in primary osteosarcoma tumors is not yet known. MET, FOS, ERBB2, and MYC are oncogenes that have been extensively investigated in osteosarcoma.
MET
MET, a receptor tyrosine kinase, was first identified and noted to be oncogenic in a transformed human osteosarcoma cell line. In the 1970s, Rhim and colleagues treated a human osteosarcoma cell line with N -methyl- N ′-nitronitrosoguanidine (MNNG), a chemical carcinogen. MNNG treatment resulted in a morphologic change and the acquisition of in vivo tumor-forming capacity. Subsequent studies have shown that the transforming event is formation of a fusion oncogene, TRP-MET, which leads to high levels of MET expression.
MET is highly expressed in human osteosarcomas and osteosarcoma cell lines. The exact proportion of osteosarcomas that have high MET expression varies, depending on the detection method used. By Western blotting, 60% to 88% of osteosarcoma tumor specimens and 100% of osteosarcoma cell lines display high levels of MET protein. Immunohistochemical analysis yields more variable results, with 35% to 75% of osteosarcomas highly expressing MET. The quantitative PCR assay also demonstrates elevated MET expression in all osteosarcoma tumor specimens and cell lines studied. Of note, the benign bone tumors osteoblastoma and nonossifying fibroma do not express MET but chondroblastoma and giant cell tumors do express MET. When primary and metastatic tumors from the same patient are compared, metastatic tumors have higher MET expression than the primary tumors, suggesting a role for MET in osteosarcoma metastasis. The mechanism of elevated MET expression in osteosarcoma is not known. When evaluated, MET gene amplification does not appear to be present in osteosarcoma.
Functional studies in cell lines have contributed to the understanding of MET-transforming roles in osteosarcoma. Expression of MET carrying an activating mutation or overexpression of MET by lentiviral transfection transforms normal osteoblasts. Despite a high level of MET expression, MET is not activated in the absence of its ligand, hepatocyte growth factor (HGF). An autocrine-paracrine loop may cause MET activation in human osteosarcomas in vivo. Addition of HGF to MET-expressing osteosarcoma cell lines results in MET activation, activation of downstream signaling intermediates MAPK and AKT, and increased invasive growth. MET-HGF interaction in other model systems induces proliferation and enhances motility, cellular changes that frequently precede metastasis.
As reviewed by Trusolino and Comoglio, the MET receptor, and HGF, turn on cellular programs that lead to invasive growth. Downstream mediators of MET activity include cadherins, integrins, and metalloproteinases. The ultimate outcome of MET activation includes loss of cell-cell adhesion, increased cell motility, and extracellular matrix degradation. Physiologic requirements for invasive growth include organogenesis, tissue regeneration, and wound healing. Inappropriate activation in cancer cells leads to tissue invasion and metastasis.
FOS
FOS was initially identified as osteosarcomagenic through investigation of the FBJ murine virus. Mice inoculated with FBJ murine virus develop osteosarcomas that are pathologically similar to human tumors. The viral protein responsible for transformation after infection with FBJ murine virus has almost complete homology with the murine c-Fos protein expressed from the murine Fos gene. Transfection of a functional, highly expressed FOS gene transforms fibroblasts.
Further evidence that c-Fos overexpression can induce osteosarcomas has been provided by transgenic mouse models. Transgenic mice in which Fos is expressed from a major histocompatibility complex (MHC) class I promoter develop osteosarcoma with 100% penetrance. In these mice, Fos is highly expressed in both osteoblasts and osteosarcomas. Transgenic mice in which the gene is expressed from a different universally expressed promoter, human metallothionein, also develop osteosarcomas but at a much lower frequency, suggesting that there is a dose-response relationship between the extent of FOS overexpression and the development of osteosarcoma.
FOS is overexpressed in human osteosarcoma tumor specimens. By immunohistochemical analysis, 50% to 60% of human osteosarcomas express high levels of FOS protein. Only 20% of benign bone tumors have immunohistochemical positivity for FOS, and the level of FOS immunohistochemical staining in benign tumors is not as high as in osteosarcomas. When compared with normal tissues and nonosteosarcomatous lesions, FOS expression, as assessed by immunohistochemistry, is 150%. FOS RNA levels are increased in 40% of osteosarcomas. One study has found an association between FOS expression and the occurrence of metastasis or recurrence, but another did not.
Although it is known that FOS, a member of the AP1 transcription factor complex, is an important mediator between extracellular signaling and transcriptional activation in normal bone development, it is not known exactly how FOS overexpression contributes to osteosarcomagenesis. Induction of Fos expression in osteoblasts alters CCND1 levels and increases entry into the S phase. Wagner has noted that AP1 complex activity is regulated by vitamin D, transforming growth factor β, and parathyroid hormone, all of which are important factors influencing bone formation, growth, and healing. In particular, FOS appears to play an essential role in enchondral ossification. Fos null mice have significant osteopetrosis, suggesting that FOS activity is required for normal osteoclast function.
Insulin-like Growth Factor
As noted earlier (see “ Etiology ”), there is epidemiologic evidence for a link between normal bone growth and osteosarcoma, including the coincidence of osteosarcoma peak incidence with maximal linear growth in adolescence and the higher rate of osteosarcoma in larger dog breeds. The observed epidemiologic links between osteosarcoma and bone growth could have alternate explanations. Osteosarcoma in large-breed dogs could be caused by the underlying genetics of breed founders rather than by rates of bone growth. Nevertheless, as central mediators of normal linear bone growth and cellular proliferation in general, insulin-like growth factor 1 (IGF-1) and its receptor, insulin-like growth factor 1 receptor (IGF1R), have been investigated in osteosarcoma. In vitro and in vivo data have supported a possible role for IGF-1 and IGF1R in osteosarcoma proliferation and invasion.
IGF-1 and IGF1R are members of the family of insulin-like growth factors and insulin-like growth factor receptors. The other ligands in the IGF-1 family are IGF-2 and insulin. There are three tyrosine kinase receptors in the family, IGF1R, IGF2R, and the insulin receptor. IGF1R is the primary target of IGF-1 and the most thoroughly studied insulin-like growth factor receptor in osteosarcoma. IGF2R also appears to be highly expressed across a number of osteosarcoma cell lines. Other key modulators of IGF1R activity are the insulin-like growth factor binding proteins (IGFBPs). Knockout mice lacking IGF-1 or IGF1R are approximately half the size of normal mice and have delayed formation of their ossification centers. Several humans with fetal growth restriction and postnatal short stature were found to have mutations in IGF1R.
Competitive binding assays, affinity labeling experiments, and Northern and Western blot studies have demonstrated expression of IGF1R by osteosarcoma cell lines. IGF-1 expression is variable across osteosarcoma cell lines. Although less thoroughly studied than IGF-1, IGF-2 appears to be expressed by osteosarcoma cells. There has been limited investigation into IGFBPs in osteosarcoma, but one study has shown that IGFBP3 transcription is augmented in MG-63 osteosarcoma cells in response to IGF-1.
In osteosarcoma cell lines, IGF1R is not activated in the absence of its ligand. Addition of IGF-1 to osteosarcoma cells in culture results in IGF-1R activation, activation of IGF-1R downstream signaling, including AKT and ERK activation, and increased proliferation One study has shown that IGF-2 is also mitogenic for osteosarcoma cell lines and that this effect is mediated by binding to IGF2R.
Multiple mechanisms of IGF1R inhibition in vitro and in vivo suppress osteosarcoma growth and invasion. In a number of different osteosarcoma cell lines, antibodies to IGF1R decrease osteosarcoma proliferation in a dose-dependent manner. In some cases, antibody treatment causes 100% inhibition of IGF-1–induced proliferation. Forced expression of IGFBP5 or IGFBP3 by genetic or chemical mechanisms results in decreased IGF-1 expression, increased expression of differentiation markers, and decreased proliferation of osteosarcoma cells. In vivo interruption of growth hormone by hypophysectomy or growth hormone–releasing hormone antagonism leads to a dramatic decrease in osteosarcoma xenograft tumor size and metastasis.
IGF-1 and IGFBP3 levels in patients with osteosarcoma are different than established age- and gender-matched normal values, with 68% of patients having low IGF-1 levels. IGF-1 and IGFBP3 levels are not correlated with clinical variables such as metastasis and outcome. Phase I studies of octreotide pamoate, a growth hormone antagonist, in patients with osteosarcoma have shown a 50% decrease in serum IGF-1 levels. However, there were no clinical responses to octreotide pamoate. There are several possible explanations that could reconcile these clinical findings, with evidence suggesting an important role for the IGF axis in osteosarcoma. The behavior of IGF-1 and IGF1R in osteosarcoma models may not accurately represent osteosarcoma in patients. More likely, IGF-1 may act in a paracrine rather than endocrine manner, so that IGF-1 serum levels are not representative of the tumor milieu. Alternatively, IGF1R activation in osteosarcoma in vivo may be mediated by IGF-2 or the extent of IGF1R activation may not be determined by ligand concentration. Finally, osteosarcoma biology is likely to be heterogeneous, with the IGF axis being important in some tumors but not in others. Early phase IGF1R antibody trials included an insufficient number of osteosarcoma patients to determine whether these drugs are effective in osteosarcoma. It is not clear whether IGF1R antibodies will continue to be developed given lack of activity of these drugs in carcinomas. IGF1R kinase inhibitors continue to be developed.
ERBB2
The ERBB2 protooncogene encodes the human epidermal growth factor receptor 2, a receptor tyrosine kinase in the epidermal growth factor receptor family. ERBB2 is amplified in 20% of breast cancers, and ERBB2 protein is expressed in a similar proportion of breast cancers. In breast cancer, ERBB2 amplification or ERBB2 expression is associated with a poorer prognosis. Trastuzumab, an anti-HER2 therapeutic antibody, improves survival in patients with ERBB2 -amplified tumors.
ERBB2 protein levels are increased in 30% to 40% of osteosarcomas. Initial studies raised the possibility of an association between ERBB2 expression and a worse prognosis, but this finding was not confirmed in later studies. Some investigators have found evidence of ERBB2 gene amplification in osteosarcoma, but others have not. As discussed later (see “ Treatment ”), these findings led to a COG study, AOST 0121, that evaluated the feasibility of adding trastuzumab to multiagent chemotherapy. Eligible patients were those with high-risk metastatic osteosarcoma whose tumors were ERBB2 positive. Outcome in ERBB2-positive patients was similar to that of historical controls, suggesting limited activity of trastuzumab in ERBB2-positive osteosarcoma.
MYC
The MYC gene, located at 8q24.1 is amplified in 7% to 10% of osteosarcomas when assessed by Southern blotting. The more sensitive technique, array CGH, detects MYC amplification in 44% of osteosarcomas. MYC mRNA and protein levels are increased in a similar proportion of osteosarcomas, and MYC expression might be correlated with metastasis. MYC, when complexed with MAX, activates the transcription of a number of genes, perhaps as many as 15% of all genes. Downstream effects of MYC transcriptional activation include proliferation, cell growth, inhibition of differentiation, and apoptosis. Exactly how MYC regulates these diverse cell processes and how MYC amplification leads to cancer is an area of active investigation. One percent of mice with conditional Myc overexpression from the EµSRa promoter develop osteosarcoma. With the recent development of BET bromodomain inhibitors, MYC has become a potential therapeutic target.
Determinants of Metastasis
One of the defining features of osteosarcoma is its tendency to metastasize early, hence the need for systemic chemotherapy in all patients. Metastasis is a complicated process that involves motility, migration, degradation of extracellular matrix, extravasation, survival during transit through vasculature, invasion, and growth. Given the complicated nature of the process, many factors are likely to play a role in metastases, but it is difficult to decipher the specific contribution of each. Several of the factors that have been more extensively studied in the context of osteosarcoma will be discussed.
Chemokine ligand 12 (CXCL12) is a cytokine-like protein expressed on the surface of vascular endothelial cells. Through binding to its chemokine receptor, CXCR4, it plays a role in cytoskeleton rearrangement, adhesion to endothelial cells, and chemotaxis. Involvement of the CXCR4/CXCL12 pathway has been implicated in the metastatic potential of several cancers, including rhabdomyosarcoma and lymphoma. CXCR4 mRNA has been shown to be expressed in osteosarcoma and associated with the presence of metastases at the time of diagnosis. In vitro assays have shown that migration of osteosarcoma cells expressing CXCR4 follows an CXCL12 gradient and that their adhesion to endothelial and bone marrow stromal cells is promoted by CXCL12 treatment. This study also provided a rationale for the propensity of osteosarcoma to metastasize to the lung, where CXCL12 concentration is high, and demonstrated prevention of pulmonary metastasis in a murine model by the administration of a CXCR4 inhibitor, suggesting molecular strategies inhibiting this axis as a therapeutic target.
Ezrin is a membrane-cytoskeleton linker protein that allows direct cellular interactions with the microenvironment, facilitating signal transduction through growth factor receptors and adhesion molecules, thereby regulating cell migration and metastasis, among other processes. In an orthotopic model of murine osteosarcoma, ezrin expression was threefold higher in the more aggressive K7M2 cell line, which correlated with its metastatic potential when compared with the less aggressive K12 cell line. In follow-up experiments, ezrin expression was found to provide an early survival advantage for pulmonary metastatic osteosarcoma, which is in part mediated by AKT. Ezrin exists in phosphorylated (active) and unphosphorylated (inactive) states. Establishment of osteosarcoma metastases in a mouse model is dependent on fluctuation between the active and inactive forms of ezrin. Ezrin phosphorylation at threonine 567 is mediated in part by protein kinase C in osteosarcoma cell lines. The identification of protein kinase C as a mediator of ezrin phosphorylation and the discovery of small molecules that inhibit ezrin phosphorylation make ezrin a potential therapeutic target for osteosarcoma. A significant correlation between high ezrin expression and poor outcome in osteosarcoma has been shown, with shorter DFS and higher risk for metastatic relapse, supporting the animal model data.
Overexpression of MET and its ligand hepatocyte growth factor, in osteosarcoma cell lines suggests a role for MET in its metastatic phenotype. Binding of the ligand to the MET ligand receptor stimulates both cell proliferation and motility, features associated with metastasis. In a study of 17 osteosarcoma tumor samples, 60% expressed the MET ligand receptor at high levels. When primary and metastatic tumors from the same patient are compared, metastatic tumors have higher MET expression than the primary tumors, indicating its role in potentiating the metastatic phenotype of osteosarcoma.
FAS is a transmembrane receptor that activates the extrinsic cell death pathway on stimulation by its ligand, FASL. There is increasing evidence that a variety of tumor types that escape from apoptosis may enhance the ability of tumor cells to metastasize. Studies in an osteosarcoma murine xenograft model with a high rate of pulmonary metastasis have suggested that decreased FAS expression enhances osteosarcoma metastasis. Therapies enhancing FAS expression inhibit the development of metastases in this model system. However, evidence that perturbations in FAS are a generalized mechanism of enhanced metastatic potential in osteosarcoma is limited.
The SAOS2 LM6 cell line was produced by serially passaging a human osteosarcoma cell line, SAOS2, through immunodeficient mice until pulmonary metastasis developed reliably and rapidly after tail vein injections. FAS protein and mRNA levels are lower in the SAOS2 LM6 metastasis–producing cell line compared with the seldom-metastasizing parental cell line SAOS2. Furthermore, when SAOS2 LM6 FAS expression is increased through transfection, the number and size of pulmonary metastases developing after tail vein injection decrease. FAS expression can also be induced by interleukin-12 (IL-12). Intrapulmonary aerosolized IL-12 gene therapy after SAOS2 LM6 tail vein injection increases FAS expression in metastatic tumors and inhibits the development of pulmonary metastases. Interestingly, ifosfamide and IL-12 display a synergistic relationship when used together in this model system, possibly because of ifosfamide-induced expression of FASL.
FAS expression has been evaluated in human osteosarcoma pulmonary metastases from 28 patients. FAS was not expressed in 60% of the tumors, and the remaining 40% had only weak FAS expression. Further study is needed to confirm that decreased FAS expression contributes to the metastatic potential of osteosarcoma occurring in humans and in osteosarcoma models other than the SAOS2 LM6 model.
Other factors that likely play a role in osteosarcoma metastases include expression of matrix metalloproteinases and other degradative enzymes. The expression of these enzymes is necessary to degrade the extracellular matrix, permitting extravasation into the vasculature.
Biologic Markers of Response to Chemotherapy
The response of osteosarcoma to chemotherapy is evident in the change in patient survival resulting from its administration. Defining “response” is somewhat more difficult. Chemotherapy response in osteosarcoma is typically used to refer to the degree of necrosis in the definitive surgical resection specimen. The degree of necrosis seen in that specimen is a consistent predictor of patient survival across studies, as described elsewhere in this chapter. In the original description of the grading system, a grade I response was considered indicative of no histologically visible response to chemotherapy. Subsequently, this was specified as being less than 50% necrosis, because untreated osteosarcomas could have as much as 25% spontaneous necrosis. It was therefore believed that necrosis more than double this amount would be attributable to a chemotherapy effect. Numerous biologic studies have been performed correlating a particular marker with the necrosis grading. In some of these studies, the necrosis grading was used as a surrogate for EFS, with the anticipation that these will correlate. The advantage of necrosis grading in this context is that the data are available much sooner than mature EFS data. In other contexts, the necrosis grading is used as a marker of chemotherapy response. Complicating these analyses, the standard of care in osteosarcoma is multiagent chemotherapy. Factors that only affect responsiveness to a single agent may not be predictive in the context of multiagent chemotherapy. Given these limitations, relating a genetic alteration to necrosis grading may provide a more robust relationship with chemotherapy response than survival, because survival may be influenced by many factors other than chemotherapy response. Similarly, necrosis grading may be a better marker of response than radiographic response in osteosarcoma, because the mineralized matrix of an osteosarcoma prevents radiographically visible shrinkage of the tumor.
Because chemotherapy has had a dramatic effect on the outcome of osteosarcoma, it is perhaps intuitive to assume that genetic alterations that produce drug resistance would be associated with inferior chemotherapy response and patient survival. Along these lines, perhaps the most extensively studied prognostic marker is the expression of P-glycoprotein (PGP) or multidrug resistance protein 1 (MDR1). This is a transmembrane adenosine triphosphate–dependent efflux pump protein encoded by the multidrug resistance gene ( ABCB1, formerly MDR1 ), which is responsible for the efflux of numerous chemotherapeutic agents from the malignant cell. In the context of osteosarcoma, the most important drug that can be effluxed by MDR1 is doxorubicin, although etoposide is also a substrate. Immunohistochemistry and reverse-transcriptase (RT)-PCR quantification studies have explored MDR1 and ABCB1 expression in osteosarcoma and demonstrated overexpression of MDR1 in 23% to 45% of tumors, with decreased survival reported for patients with MDR1-positive tumors in initial studies. Of interest, although many of these studies demonstrated a correlation with outcome, several key studies did not demonstrate a relationship with the necrosis grade. This suggests that use of necrosis grading as an early marker of outcome may not always be appropriate in the context of some biologic markers. A meta-analysis has proposed that MDR1 is associated with an increased risk for disease progression. Although the early literature suggested that MDR1 may be a marker of drug resistance and aggressiveness in osteosarcoma, this finding has not been verified consistently across studies. In addition, a prospective clinicopathologic cooperative group study has concluded that there is no correlation between MDR1 expression and percentage of osteosarcoma tumor necrosis after induction chemotherapy or EFS in localized osteosarcoma. Overexpression of ABCB1 has also been explored with regard to its prognostic relevance. Although a small pilot study has shown a trend toward a worse outcome in patients exhibiting high levels of ABCB1 expression, a larger prospective investigation did not identify a correlation between ABCB1 messenger RNA expression and prognosis in osteosarcoma patients, with patients with either very low or very high levels of ABCB1 having a relatively poor outcome.
High-dose methotrexate (HDMTX) with leucovorin rescue is a major component of current protocols for the treatment of osteosarcoma. HDMTX is vastly more effective than conventional dose methotrexate in the treatment of osteosarcoma, a finding that is not observed in other malignancies treated with MTX, implying a mechanism of intrinsic MTX resistance within osteosarcoma tumor cells. In experimental systems, resistance to methotrexate can occur through a variety of mechanisms, including impaired intracellular transport of the drug via the reduced folate carrier, upregulation of dihydrofolate reductase, and diminished intracellular retention caused by decreased polyglutamylation. Studies have demonstrated that impairment of drug influx as a result of decreased expression and mutations in the reduced folate carrier gene is the major basis of intrinsic resistance; 65% of osteosarcoma tumor samples were found to have decreased reduced folate carrier expression at the time of initial biopsy. In contrast, dihydrofolate reductase overexpression was seen relatively infrequently at initial biopsy, in only 10% of tumor samples, compared with 62% of the tumors examined at the time of definitive surgery or relapse, suggesting dihydrofolate reductase overexpression as the major mechanism of acquired MTX resistance in osteosarcoma.
In vitro induction of HMGB1 expression in osteosarcoma cells prevented apoptosis. HMGB1 is a chromatin-binding nuclear protein that may be involved in chemotherapy resistance in other cancers. Evaluation of HMBG1 in human osteosarcoma samples has not been performed. Other potential mechanisms of drug resistance include alterations in multidrug resistance protein expression, topoisomerase II, glutathione S -transferases, DNA repair, DNA damage response, drug metabolism or inactivation, and reduced intracellular delivery. Few studies have been performed in osteosarcoma to indicate whether these processes play a role in defining chemotherapy response.
In other studies, necrosis after chemotherapy has been used as a marker of response, and investigators have attempted to identify genetic signatures predictive of response. In each of these studies, a large number of genes were required to define a profile of response. The studies reported using 60 genes, 104 genes, and 45 genes to distinguish between responders and nonresponders. This may be related to the small sample sizes used in each of these studies. Alternatively, the lack of robust clustering may be caused by the heterogeneity of osteosarcoma itself. Lack of robust clustering on gene expression profiling appears to be a property of non–translocation–associated sarcomas, a result of their high levels of genetic complexity.
Translational Science
Bone metabolic pathways are of interest in osteosarcoma given that this is a tumor arising from osteoblasts and osteoblast precursors. Zoledronate is a nitrogen-containing bisphosphonate and the most potent of its class. It inhibits the development of osteolytic and osteoblastic bone lesions. Preclinical models have shown cytotoxicity against osteosarcoma cell lines. In vivo models in mice inoculated intravenously with the spontaneous murine osteosarcoma cell line POS-1 have shown prolonged survival when treated with single-agent zoledronate. Moreover, combination with ifosfamide has shown more activity than either agent alone in preventing tumor recurrence, improving tissue repair, and increasing bone formation. Bisphosphonates may also improve bone integrity after limb salvage surgery.
Receptor activator of nuclear factor κB ligand (RANKL, also known as tumor necrosis factor ligand superfamily member 11 [TNFSF11]) and its receptor RANK have the physiologic function of regulating bone turnover. In response to RANKL-RANK binding, osteoclast precursors differentiate and become activated resulting in bone resorption. An antibody against RANKL, denosumab, has been approved by the U.S. Food and Drug Administration (FDA) for treatment of osteoporosis. RANK is expressed in 57% of human osteosarcomas, most human osteosarcoma cell lines, and 70% of canine osteosarcomas, including two of two canine osteosarcoma skin metastases. RANKL is expressed in 75% of human osteosarcoma tumor specimens, and the strength of expression is associated with prognosis. In osteosarcoma cell lines, RANKL activates downstream signaling and modulates gene expression. Further evidence supporting activity of the RANK-RANKL pathway in osteosarcoma is provided by a transgenic mouse model of osteosarcoma. In an osteosarcoma mouse model created by expression of the SV40 T/t antigen from the osteocalcin promoter, heterozygous deletion of Prkar1a accelerates development of osteosarcoma via increased Rankl gene expression. A subset of human osteosarcomas have low PRKAR1A expression and high RANKL expression. In vivo studies of denosumab are limited by the fact that the antibody does not recognize murine or canine Rankl. To circumvent these limitations, in vivo studies have utilized other approaches to inhibit Rankl activity, including Rank gene therapy, which creates a decoy receptor for Rankl, RANK-Fc, a chimeric protein that efficiently binds Rankl, or SiRNA directed against Rankl. In a murine orthotopic OS model, Rank gene therapy decreased primary and metastatic tumor burden: mice receiving this therapy did not develop osteosarcomatous lung metastases whereas 80% of control mice developed lung metastases. A study by the same group using RANK-Fc confirmed these original findings.
The phosphatidylinositol 3′ kinase/mammalian target of rapamycin (PI3K/mTOR) pathway is a central signaling pathway contributing to proliferation and survival in many cancers. The pathway has been recognized to contribute to proliferation, drug resistance, and metastasis in osteosarcoma. Expression of mTOR and p70S6K, an indicator of mTOR activation, is present in 70% to 80% of osteosarcomas, and the level of expression has been correlated with overall and disease free survival. Activation of PI3K-AKT signaling is downstream of several biological processes involved in osteosarcoma metastasis such as Ezrin expression. Micro-RNA-221, expressed at high levels in osteosarcoma targets PTEN, increases cell survival, decreases apoptosis, and induces cisplatin resistance. The dual PI3K/mTOR inhibitor BEZ235 and the alpha selective PI3K inhibitor BYL719 induce cell cycle arrest and decrease tumor formation in vivo. The clinical trial experience with mTOR inhibition in osteosarcoma is limited, and PI3K inhibitors have not been evaluated in this disease. There have been rare but dramatic responses to mTOR inhibition in patients with advanced, refractory osteosarcoma. In a study using a sequenom assay to identify cancer-associated mutations in osteosarcoma, 3 of 89 tumors were found to have mutations in PIK3CA , the PI3K catalytic subunit. A comprehensive next-generation sequencing study of 59 osteosarcoma samples thus far presented only in abstract form, identified PI3K/mTOR pathway alterations in 25% of osteosarcomas.
GD2 is a cell surface sialic acid containing glycosphingolipid expressed normally in the central nervous system, peripheral nerves, and melanocytes. In neuroblastoma, anti-GD2 antibody given with cytokines to promote antibody-dependent cell-mediated cytotoxicity significantly improves EFS and OS. More than 90% of osteosarcomas express GD2, and the intensity of expression, in most samples, is greater than that seen in neuroblastoma. In the phase I/Ib trial of the murine anti-GD2 antibody, 14.2G2a, plus interleukin-2, one of two osteosarcoma patients treated had a complete response persisting for 8 months.
There is mixed evidence regarding the role for immunotherapy in osteosarcoma. Completed clinical trials utilizing general immune stimulation with interferon α (IFN-α) and inhaled granulocyte-macrophage colony-stimulating factor (GM-CSF) did not demonstrate significant activity of either approach. Dendritic cell therapy has shown anti-osteosarcoma activity in one mouse model, particularly when pulsed in vitro with irradiated tumor and administered in a minimal residual disease setting. However, in a clinical study of dendritic cell vaccination there was a lack of T-cell response and no evidence of clinical benefit. Immune surveillance by T cells may delay or prevent the development of metastasis, as seen in a mouse model. In a single study in a murine xenograft model, chimeric antigen receptor T cells expressing interlukin-11Rα treatment resulted in regression of pulmonary metastases.
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


