Definition of Osteoporosis
The term osteoporosis was first introduced in the nineteenth century based on histological diagnosis (“porous bone”). Osteoporosis is a “disease characterized by low bone mass and microarchitectural deterioration of bone tissue leading to enhanced bone fragility and a consequent increase in fracture incidence.” Osteoporosis can be defined either by the presence of a fragility fracture or by bone mineral density (BMD) measurement. In defining BMD criteria for osteoporosis, the World Health Organization (WHO) used as the standard the BMD of young adult women who are at the age of peak bone mass. For each standard deviation below peak bone mass (or 1 unit decrease in T-score), a woman’s risk of fracture approximately doubles. As seen in Table 117-1, a T-score <−2.5 defines osteoporosis; osteopenia (low bone mass) and normal bone mass are also defined. A BMD measurement allows early diagnosis of osteoporosis and intervention prior to fracture in older adults. In addition, women with osteopenia can be followed carefully for further bone loss. Although the original standards for definitions of osteoporosis were determined in white women, recent data indicate that the standards for men and Hispanic women are probably similar to those of white women. In addition, standards are now being developed for African-American women. However, defining osteoporosis solely by T-score does not effectively capture all patients at risk of a fracture. Greater than 50% of all hip fractures occur in those with T-scores that are better than −2.5. Failure to evaluate and treat such patients adds to the individual and societal cost and consequences of osteoporosis. Therefore, we are still faced with the challenge of improving the identification of the individual patient at risk of fracture and subsequently optimizing both prevention and treatment for the elderly.
Primary or idiopathic osteoporosis has been historically classified as postmenopausal or senile osteoporosis. Postmenopausal osteoporosis, formerly known as type I, occurs in women between 51 and 75 years of age and is related to estrogen deficiency seen with the menopausal transition. In contrast, senile osteoporosis typically occurs in persons older than 60 years of age. It affects both men and women and has a different pathophysiology, which involves reduced levels of bone turnover owing to a reduction in the numbers of bone-forming cells. Increasing evidence points to a progressive age-related alteration in stem cell physiology that favors adipogenesis and thereby reduces osteoblastogenesis and bone formation. Nevertheless, estrogen probably plays a role in the pathophysiology of senile osteoporosis as well. Secondary osteoporosis is the result of an underlying disease or medications that adversely affect bone. In this chapter, we will focus on the typical characteristics of senile osteoporosis from its pathophysiology to therapeutic approaches.
Epidemiology
In the United States, women have more than 250 000 and 500 000 hip and spine fractures per year, respectively. Men account for an additional 250 000 fractures per year; 75 000 are hip fractures. By using BMD, osteopenia is estimated to be present in 8 to 13 million men (28–47%) and in 13 to 17 million women (37–50%); osteoporosis is estimated to be present in 1 to 2 million men (3–6%) and in 4 to 6 million women (13–18%). On reaching the age of 90 years, one-third of women and one-sixth of men will suffer a hip fracture. Both women and men have a similar lifetime vertebral fracture risk of 12%.
The consequences of osteoporotic fracture include diminished quality of life, decreased functional independence, and increased morbidity and mortality. Pain and kyphosis, height loss, and other changes in body habitus resulting from vertebral compression fractures diminish quality of life in women and men. These changes lead to declines in functional status such as the inability to bathe, to dress, or to ambulate independently and may result in decrease pulmonary and gastrointestinal function. Approximately 50% of women do not fully recover prior function after hip fracture; older adults have a 20% to 25% mortality in the year following hip fracture. Men are at a higher risk of dying after a hip fracture than women. Finally, the estimated annual cost of osteoporotic fractures in the United States is more than $14 billion, which is higher than the money spent treating cardiovascular disease. Therefore, prevention and early diagnosis and treatment of osteoporosis are vital to improving the health of older adults.
Pathophysiology
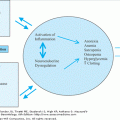
New advances in the understanding of bone physiology have elucidated an active interaction among bone cells, growth factors, and hormones responsible for the maintenance of calcium levels, skeletal structure, and resistance to trauma. Bone is not simply a mineralized structure but a complex system of cell–cell, cell–matrix, and cell–hormone interactions influenced by genetic background, lifestyle, and diet.
Bone is composed of inorganic (calcium phosphate crystals) and organic compounds (90% collagen and 10% noncollagenous proteins). Noncollagenous proteins include albumin, osteopontin, osteocalcin, α2-HS-glycoprotein, and growth factors, constituting the so-called bone matrix. The bone matrix is produced by osteoblasts and is the environment in which bone and external factors interact in a well-coordinated manner. There are two types of bone: cortical and trabecular. Cortical bone predominates in the long bones of the extremities, while trabecular bone predominates in the vertebrae and pelvis and makes up 80% of skeletal mass. While both types of bone have an active remodeling process, trabecular bone is metabolically more active than cortical bone and more acutely responsive to alterations in sex-steroid hormone status.
During childhood and adolescence, skeletal growth takes place at growth plates, areas in which cartilage proliferates and gradually undergoes calcification, resulting in new bone formation. However, bone remodeling is a lifelong process that maintains bone in order to harbor bone marrow, support the body, and protect vital organs and to provide a source of minerals. The process of remodeling replaces older, frailer bone with newer, more resistant bone in an organized manner. The end product of remodeling is the maintenance of skeletal homeostasis and the preservation of anatomical integrity. With aging or with menopausal transition, the once-coordinated mechanism of bone remodeling becomes uncoupled, leading to bone loss and increased risk of fracture.
The cells involved in bone turnover are osteoclasts, osteoblasts, and osteocytes. Osteoclasts are macrophage-like cells that secrete proteolytic enzymes and hydrogen ions that are required for removal of the deposited bone matrix. The remodeling cycle begins when osteoclast precursors interact with other marrow cells and are activated, becoming multinucleated osteoclasts, which initiates resorption (Figure 117-1). Bone resorption occurs within the resorption lacuna, a tightly sealed zone beneath the ruffled border of the osteoclast where it has attached to the bone surface. Resorption depends on acidification of this extracellular compartment leading to demineralization, and, subsequently, the organic matrix is degraded by cysteine proteases, chief of which is cathepsin K. Osteoclasts consequently create a functional extracellular lysosome, containing both an acidic environment and typical lysosomal enzymes.
Figure 117-1.
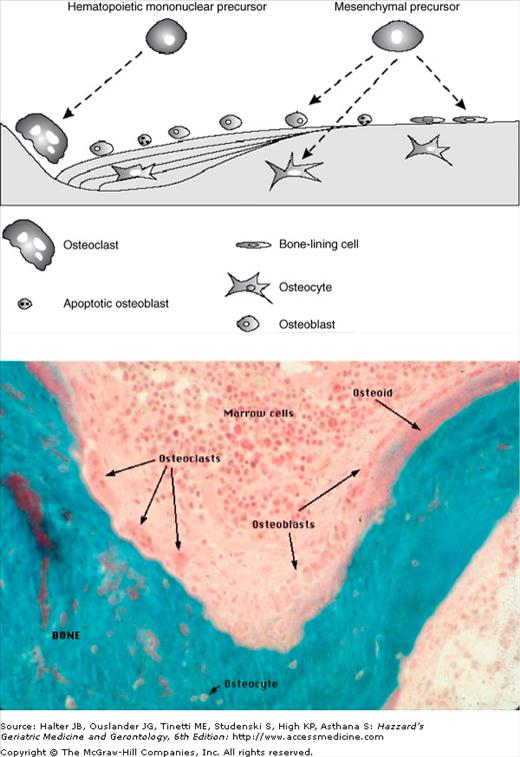
The cellular components of bone turnover. (A) Bone cell formation: After the expression of specific transcription factors, mesenchymal precursors differentiate into osteoblasts. In contrast, osteoclasts differentiate from mononuclear precursors and will act as bone-resorbing cells in the bone multicellular unit (BMU). After the completion of bone resorption, osteoclasts die by apoptosis and are replaced by active osteoblasts, which are responsible for the formation of new bone. Finally, osteoblasts end as either lining cells or as osteocytes embedded into the osteoid, or die by apoptosis. (B) Basic multicellular unit (BMU—also known as bone metabolic unit): Large multinucleated osteoclasts resorb bone on the left. Osteoblasts cascade into the resorption pit, laying down osteoid (unmineralized bone). The turquoise stain reflects mineralized bone. (From Chan GK, Duque G. Age-related bone loss: old bone, new facts. Gerontology. 2002;48:62–71 [Figure 117-1A]; and Dr. Susan M. Ott http://courses.washington.edu/bonephys [Figure 117-1B].)
In cortical bone, the resorption period lasts approximately 30 days; the final result is a resorption tunnel that will later be filled in by osteoblasts in a haversian manner. The apposition of these haversian channels takes the shape of a “cut onion,” which gives the cortical bone its typical morphology. In trabecular bone, the erosion period lasts approximately 43 days, resulting in a trench between the trabeculae. The life span of osteoclasts is approximately 2 weeks; once these complete the role as bone-resorbing cells, these undergo apoptosis or programmed cell death.
Bone homeostasis depends the intimate coupling of bone formation and bone resorption. After bone is resorbed by osteoclasts, preosteoblasts differentiate into osteoblasts and migrate to the area of excavated bone and begin to deposit osteoid, which is eventually mineralized into new bone. Osteoblasts are fibroblast-like cells derived from pluripotent mesenchymal cells and localized on periosteal surfaces. Such pluripotent stromal cells can be induced to differentiate along the osteoblastic, adipocytic, fibroblastic, or chondrocytic lineages when required. When bone integrity has to be conserved, mesenchymal stromal cells are committed toward the osteoblastic lineage. Many factors are involved in the process of osteoblastogenesis, including the bone morphogenic protein family—bone morphogenic proteins 2, 4, and 7 are potent inducers of osteoblast differentiation (Table 117-2). A transcription factor called Runx2/Cbfa1 plays a key role in osteoblast differentiation; mice lacking Runx2/Cbfa1 do not form bone. A mature osteoblast is a cuboidal cell with a large nucleus and enlarged Golgi highly enriched in alkaline phosphatase. It produces type I collagen and specialized bone-matrix proteins as osteoid, the basic protein for further bone formation and mineralization. Osteoblasts produce alkaline phosphatase, the specific function of which has yet to be determined. Nevertheless, it is used as a marker of osteoblast differentiation and activity, and indirectly as a marker of subsequent osteoclast resorption. Mice lacking functional alkaline phosphatase suffer from hypophosphatasia characterized by impaired mineralization of cartilage and bone matrix. After osteoblasts complete their bone-forming function, they face one of three fates: (1) they become embedded in the newly formed matrix, becoming osteocytes; (2) they remain on the surface of the newly formed bone and become lining cells; or (3) they undergo apoptosis (see Figure 117-1). The ultimate fate of the osteoblast is determined by hormonal changes, the presence or absence of growth factors, and the aging process in bone.
Stimulators of bone resorption | Stimulators of bone formation |
| RANK ligand | Interleukin-1, interleukin-6, interleukin-8, interleukin-11 |
| Macrophage colony-stimulating factor | Vitamin D |
| Vitamin D | Estrogen |
| Glucocorticoids | Androgen |
| Parathyroid hormone | Insulin-like growth factors |
| Tumor necrosis factor | Transforming growth factor |
| Reduced mechanical force/low gravity | Mechanical force |
| Epidermal growth factor | Fibroblast growth factors |
| Platelet-derived growth factor | Platelet-derived growth factor |
| Fibroblast growth factors | Bone morphogenetic proteins |
| Leukemia inhibitory factor | |
| Prostaglandins | |
| Thyroid hormone | |
Inhibitors of bone resorption | Inhibitors of bone formation |
| Estrogen | Sclerostin |
| Androgen | Dickkopf |
| Osteoprotegerin | |
| Interferon-γ | |
| Interleukin-4 | |
| Calcitonin |
Osteoclasts and osteoblasts belong to a temporary structure known as basic multicellular unit (see Figure 117-1). The coordinated process of bone resorption and formation by the basic multicellular unit lasts 6 to 9 months and results in newly mineralized bone. Osteoblasts are not only active as bone-forming cells, but these also play an important role in the regulation of osteoclast activity. The interaction between osteoblasts and osteoclasts requires a complex system of factors and is facilitated by integrins and cadherins. Briefly, the major osteoclast/osteoblast interaction depends on the expression by osteoclast precursors and mature osteoclasts of a membrane receptor known as receptor activator of nuclear factor-κB (RANK), which belongs to the family of tumor necrosis factor (TNF) receptors. Osteoclast differentiation, maturation, and survival depend on the activation of RANK by its cognate ligand (RANK ligand—RANKL), which is produced by osteoblasts and osteoblast precursors after exposure to different stimuli such as hormones and cytokines (Figure 117-2). Multiple other factors also act to either enhance or suppress osteoclast formation and activation and subsequent bone resorption (see Table 117-2).
Figure 117-2.
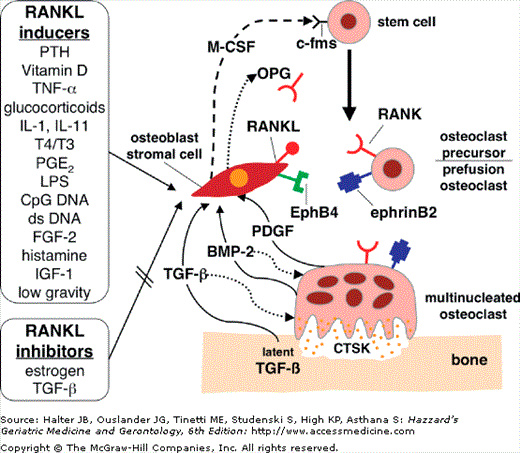
Osteoblast–osteoclast coupling and the regulation of RANK ligand expression. Osteoblast production of M-CSF and RANKL play critical roles in the differentiation and activation of osteoclasts. M-CSF acts to maintain monocytic stem cell survival, and subsequently RANKL acts to commit the cell toward osteoclast differentiation, fusion, polarization, and activation. EphB4 and ephrinB2 interact both to limit osteoclast activity and stimulate osteoblast differentiation. TGF-β acts only upon release from the extracellular matrix after osteoclastic resorption, which is mediated in large part by the excretion of CTSK. M-CSF, macrophage colony stimulating factor; RANKL, RANK ligand; TGF-β, transforming growth factor-β; BMP-2, bone morphogenetic protein-2; PDGF, platelet-derived growth factor; CTSK, cathepsin K. (Adapted from Duque G, Troen BR. Understanding the Mechanisms of Senile Osteoporosis: New Facts for a Major Geriatric Syndrome. J Am Geriatr Soc. 2008;56:935–941).
RANKL is mainly a cytoplasmic membrane-bound molecule; to a lesser extent it is secreted. Mature osteoblasts also produce a decoy receptor for RANKL called osteoprotegerin (OPG). OPG binds to RANKL in a competitive manner and prevents the interaction between RANK and RANKL, thus decreasing osteoclastogenesis and osteoclastic bone resorption and increasing osteoclast apoptosis. Recently, a new group of molecules, known as ephrins, has been identified as key players in the regulation of osteoblast/osteoclast interaction. This cellular communication is bidirectional and involves a transmembrane ligand known as ephrinB2, which is expressed by osteoclasts, and its receptor EphB4 expressed by osteoblasts (see Figure 117-2). This signaling seems to limit osteoclast activity while enhancing osteoblast differentiation. Consequently, osteoblastogenesis and osteoclastogenesis, along with corresponding bone formation and resorption, are tightly and ineluctably coupled. The differentiation and activation of both osteoblasts and osteoclasts depend critically on each other.
Osteocytes, the third group of bone cells, are the most abundant cell type in bone. Osteocytes are the focus of intense research; several functions recently described include the synthesis of matrix molecules like osteocalcin and an important role in direct communication with surface osteoblasts through molecules known as connexins. Connexins are believed to play a role in the activation and regulation of bone after physiological and mechanical stimuli. These modulate the response of bone during functional adaptation of the skeleton to mechanical forces and the need for repair of microdamage. More recently, sclerostins have been found to be secreted by osteocytes. Sclerostins inhibit osteoblast differentiation and activation, thereby regulating bone turnover and formation. The final fate of the osteocyte is unknown, but high numbers of apoptotic cells have been found in empty lacunae.
Genetics plays a role in determination of peak bone mass. Racial differences in the incidence of osteoporosis have been reported, including a lower relative risk of fractures and higher peak bone mass in African-American women as compared with white women. No one gene, gene product, or polymorphism has yet been credibly identified to account for the variance seen in BMD in specific geographic areas. Several environmental factors, such as diet, topography, and yearly sunlight exposure, almost certainly interact with genetic predisposition to explain variance seen in periosteal expansion before puberty and in trabecular number and thickness and periosteal–endosteal remodeling during aging. Several recently described candidate genes for determination of peak bone mass are the vitamin D receptor, the peroxisome proliferator activator gamma, and the low-density lipoprotein receptor-related protein 5. All these polymorphisms have been associated with different levels of peak of bone mass and predisposition to fractures in the adulthood. More generally, there does appear to be a familial predisposition to osteoporotic fracture. Therefore, the risk of fracture is increased if an immediate family member (most typically a mother or sister) has experienced an osteoporotic fracture.
Approximately 95% of peak adult bone mass is gained by the end of puberty. The level of peak bone mass attained and the subsequent rate of bone loss are the primary factors that determine an individual’s bone mass in early and late adulthood. Initial bone formation does not require a mechanical stimulus, but further appositional and endochondral growth is dependent on the mechanical forces generated by the muscles. The magnitude of this loading is directly related to body mass and physical activity. There is some evidence that after mechanical load, microfractures may occur in bone, with subsequent activation of interleukins (ILs) and growth factors and stimulation of bone turnover. Osteocytes appear to be the critical mechanosensors in bone and, therefore, play an important role in this process. The decreased physical activity and immobility that are commonly seen in the elderly result in diminished mechanical forces upon the skeleton, which explains, in part, the age-related loss in mineral density.
Local factors are important in the regulation of bone turnover and in the interaction between bone matrix and systemic factors and hormones (see Table 117-2 and Figure 117-2). The skeleton responds to mechanical forces by several regulatory mechanisms including release of cytokines, such as macrophage colony-stimulating factor (M-CSF) and granulocyte colony-stimulating factor, which regulate cell differentiation. Mediators and regulators of cell–cell interaction include insulin-like growth factor (IGF)-1 and IGF-2, parathyroid hormone (PTH)-related peptide, IL-1 and IL-6, and TNF-α. The response to sex-steroid hormones is largely mediated by TNF-α, IL-6, IL-1, and prostaglandins. Although high levels of these factors are necessary for osteoblast–osteoclast regulation and pathogenesis of osteoporosis, their usually stable systemic levels suggest that alterations in local secretion and concentration are critical to bone physiology. These local factors largely determine the activation or inhibition of bone cells, cell recruitment and cell differentiation, and life span.
A number of systemic hormones affect bone metabolism, including vitamin D, PTH, calcitonin, and sex-steroid hormones (estrogens and androgens) (see Table 117-2). The major effect of vitamin D is to maintain calcium homeostasis by increasing the efficiency of the small intestine in absorbing dietary calcium. Vitamin D also plays a role in both bone resorption by inducing RANKL expression by osteoblasts, thereby inducing osteoclast differentiation and activation, and bone formation by stimulating osteoblastogenesis and inhibiting apoptosis of mature osteoblasts. Hypovitaminosis D, which is widespread in the elderly, is associated with lower BMD, greater falls, and more osteoporotic fractures.
PTH is secreted by the parathyroid glands through a calcium sensor mechanism. When calcium levels decrease, PTH is released and exerts its function in two major target tissues: kidney and bone. In the kidney, PTH acts on the proximal tubule to reduce PO4 resorption and to increase the activity of 1-α-hydroxylase, the enzyme that converts 25(OH) vitamin D to 1,25(OH)2D3, the active form of vitamin D. In bone, PTH increases osteoclast-induced bone resorption by inducing RANKL expression and subsequent signaling via RANK. Hypovitaminosis D is often, but not always, accompanied by elevated PTH—secondary hyperparathyroidism. Acute and cyclical exposure to PTH in bone has an antiapoptotic as well as anabolic effect on osteoblasts. This is the basis for using PTH to treat severe osteoporosis (see below). Calcitonin is a hormone secreted by thyroidal C cells in mammals. Its main biologic effect is the inhibition of osteoclastic bone resorption. In vitro and in vivo studies in animals demonstrate that calcitonin causes the osteoclast to shrink and retract from the bone surface, which decreases its bone-resorbing activity and enhances bone formation by osteoblasts.
Sex-steroid hormones play a variety of roles in bone turnover. Although some aspects of its effects remain unclear, estrogen increases the level of OPG, inhibiting osteoclastogenesis. Estrogen also induces osteoclast apoptosis and regulates the action of IL-1, interleukin-1 receptor antagonist (IL-1Ra), IL-6, and TNF-α, and their binding proteins and receptors. Declining estrogen levels lead to increased expression of IL-1, IL-6, and TNF-α, all of which enhance bone resorption. In response to diminished estrogen, osteoblasts produce more RANKL and less OPG, which induces RANKL–RANK interaction and signaling, further stimulating osteoclast differentiation and activation. Since estrogen increases osteoblast differentiation and decreases osteoblast apoptosis, bone formation declines at the time of menopause. Overall, there is a high turnover state with predominant bone resorption, which results in bone loss and susceptibility to fractures. Androgens play an important role in the formation of adolescent bone by regulating cytokines in the bone matrix. The effect of progesterone on bone seems to be indirect and limited, through its regulation of calcitonin secretion and thus bone resorption.
Women are at higher risk of osteoporosis because women have lower peak bone mass than men and experience accelerated bone loss during menopause, as described above. Histomorphometric data on the skeletal changes associated with postmenopausal bone loss show an increase in bone turnover in both cancellous and cortical bones. Biochemical markers also support high bone resorption after menopause. These markers return to normal with estrogen replacement. Trabecular bone is affected earlier in menopause than cortical bone because it is more metabolically active. Thus, rapid bone loss is seen primarily in the spine (3% per year) for approximately 5 years after menopause. Subsequently, there is a slower rate of bone loss that is more generalized (approximately 0.5% per year at many sites). A consistent finding in untreated postmenopausal women is a reduction in wall width of bone, indicating reduction in osteoblast activity. Although this could be related to the loss of the antiapoptotic effect of estrogen on osteoblasts, studies are inconclusive.
Age-related bone loss is a complex phenomenon, with many factors involved in its pathogenesis (Figures 117-3 and 117-4). As an individual ages, distinct changes take place in trabecular bone, cortical bone, and bone marrow. The onset and triggers of age-related bone loss are not well defined, but densitometric studies show a slow and progressive decline in BMD after the third decade of approximately 0.5% per year, even though serum levels of estrogens are still within the normal range. With aging, osteoblastogenesis decreases, resulting in lower numbers of osteoblast precursors and increasing bone marrow adiposity (see Figure 117-3). The bone marrow of a young individual is virtually devoid of adipocytes. In older adults, however, adipose deposits may occupy up to 90% of the bone marrow cavity.
Figure 117-3.
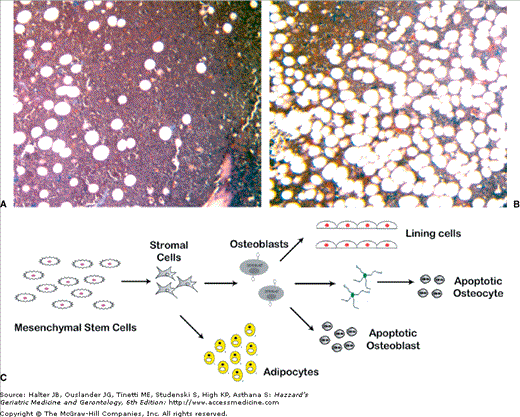
Cellular changes in senile osteoporosis. The panel on the right shows the much higher levels of bone marrow fat in the iliac crest of an 85-yr-old subject (A) compared with a 38-yr-old one in the panel on the left (B). There is also a marked reduction in the amount of hematopoietic tissue. (C) Changes in the confluence of mesenchymal stem cells (MSC) accompanied by a reduction in osteoblastogenesis result in the formation of fewer active osteoblasts in the bone multicellular unit. In addition, increasing levels of adipogenic differentiation leads to smaller numbers of differentiated osteoblasts. Finally, increasing osteoblast apoptosis reduces the number of active osteoblasts in the bone multicellular units.
Figure 117-4.
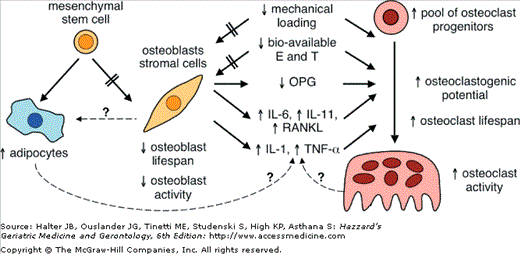
Changes in osteoblast–osteoclast interactions with aging. Aging predisposes to more adipogenesis and less osteoblastogenesis. In addition, transdifferentiation from osteoblasts to adipocytes may occur. Reduced physical activity/mechanical loading and decreased levels of bioavailable estradiol and testosterone exert diminished effects on osteoblasts (depicted by the arrows with the hatches), resulting in decreased osteoblast secretion of osteoprotegerin (OPG) and increased expression and secretion of RANKL, IL-1, IL-6, IL-11, and TNF-α. In turn, these compounds directly stimulate greater osteoclast formation and activity. The reduced OPG also permits greater binding of RANKL to RANK, which further facilitates increased osteoclastogenesis and resorption. (Adapted from Troen BR. Molecular mechanisms underlying osteoclast formation and activation. Exp Gerontol. 2003;38(6):605–614.)
Although factors that are involved in osteoblast–adipocyte differentiation remain unclear, it is known that adipocytes can transdifferentiate into osteoblasts and osteoblasts can be induced to transdifferentiate into adipocytes (see Figure 117-4). It has been postulated that pluripotent mesenchymal cells within the bone marrow stroma are, by default, programmed to differentiate into adipocytes, but the presence of specific osteogenic factors in the bone marrow induces osteoblastic differentiation. If this hypothesis is proven true, then it could offer the potential for novel therapeutic approaches to osteoporosis in older adults. In addition, osteoblast apoptosis increases with aging. Histomorphometric data demonstrate that 50% to 70% of the osteoblasts present at the remodeling site cannot be accounted for after enumeration of lining cells and osteocytes. The discrepancy in osteoblast numbers is believed to be a consequence of osteoblast apoptosis. This phenomenon may account for the significant reduction in bone formation that is associated with aging. In men and women, the early changes associated with age-related bone loss are similar, as described above. However, women also experience accelerated bone loss of approximately 3% to 5% per year during menopause. In men, the decline in bone mass is gradual until very late in life, when the risk for fractures increases rapidly. Concomitant with the changes in osteoblast and adipocyte formation, multiple factors enhance osteoclastogenesis and bone resorption (see Figure 117-4). In particular, the interactions between osteoblast and osteoclasts, which are crucial to the dynamic equilibrium that maintains healthy bone, are altered. Consequently, the combination of decreased bone formation and increased bone resorption leads to diminished BMD, poorer bone structure and quality, and, ultimately, enhanced fragility and fractures.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


