Oncology and Aging: General Principles: Introduction
This chapter discusses many of the general relationships of oncology and aging. It focuses on the epidemiological, basic etiological, and biological relationships between the processes of aging and neoplasia, and on the generalizable aspects of management of malignant disease in the elderly patient. This chapter discusses clinical management of individual malignancies only as an example of general principles. The approach to specific malignancies is covered in subsequent chapters related to the appropriate organ system.
It is now well recognized that cancer is a major problem for elderly individuals. It is the second leading cause of death after heart disease in the United States, and age is the single most important risk factor for developing cancer. Approximately 60% of all newly diagnosed malignant tumors and 70% of all cancer deaths occur in persons 65 years or older according to the NCI Surveillance. As illustrated in Figure 94-1, the total cancer incidence rises progressively through the middle years and then falls off in the later years. However, the age-specific cancer incidence rises progressively throughout the age range. Thus, while the rate of increase diminishes somewhat in the oldest age groups, and the rate actually falls slightly in the very oldest (perhaps a survivor effect), the overall risk for developing cancer is certainly greatest in the later years. Because the number of people in this country older than age 65 years is rising rapidly and the oldest of the old, that is, those older than age 85 years, are increasing at the greatest rate, geriatricians, generalists, and internists will be encountering increasing numbers of elderly individuals with cancer in their practices.
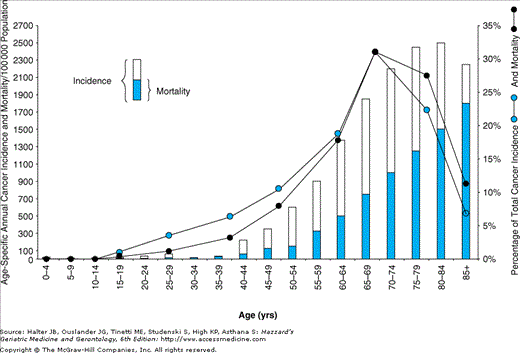
The median age range for diagnosis for most major tumors, common to both men and women, is 68 to 74 years; the median age range at death is 70 to 79 years. The overall pattern for the incidence of age-specific cancer shows a rise with age; overall, 60% of cancers occur in those age 65 years or older (Table 94-1). This is not uniform for individual cancers and in some malignancies, there is an apparent decrease in incidence in people older than age 80 years. This may be a result of a number of factors, including underreporting or natural selection, which would allow the less-cancer-prone population to survive. However, cohort effects may have the most significant impact. For example, age-specific annual cancer incidence rates from the SEER Program indicate a fall in incidence in the oldest age groups for both prostate and lung cancer. This changes when the data are corrected for certain known risk factors. For prostate cancer, when only men are considered in the base population at risk, the incidence continues to rise into the oldest age groups. For lung cancer, an apparent decrease in lung cancer incidence in the older age groups might be explained by a smaller high-risk population because of decreased prevalence of smoking in the older age groups. When data derived from the Lung Cancer Early Detection Project for annual cancer incidence in male smokers older than the age of 45 are used, one notes a continuing increase into advanced age. There is little change in the case of colorectal cancer, because the entire population appears to be at risk. For women with breast cancer, data indicate an incidence that continues to rise slowly into advanced age. It has been suggested that data from the most recent survey showing a decrease in breast cancer risk at older ages (>75 years) may be an artifact of recent increases in breast cancer screening in the United States. For other gynecologic malignancies, there does appear to be a decrease, perhaps because of different interactions of hormonal status and neoplasia in hormonally responsive target organs.
CANCER | % 65+ |
|---|---|
Prostate | 63.8 |
Colon | 70.2 |
Pancreas | 69.2 |
Urinary bladder | 72.2 |
Stomach | 65.5 |
Rectum | 57.0 |
Lung and bronchus | 67.8 |
Leukemias | 54.3 |
Corpus uteri | 45.3 |
Non-Hodgkin’s lymphoma | 54.3 |
Breast | 42.3 |
Ovary | 47.0 |
All Cancers Combined | 55.8 |
Other types of patterns in age-specific incidence may also be seen. For example, Hodgkin’s disease has a distinct bimodal distribution in incidence with a peak in the early years and another peak after late middle age. This has led to the suggestion that there actually may be two different diseases involved, one in the young individual and one in the older one, but that they assume similar morphologic features, and so with current technologies we are unable to tell them apart. This impression is further substantiated by the markedly different response to treatment in younger and older groups of individuals with this disease. On the other hand, the most common leukemias and lymphomas in elderly patients are those derived from the B-lymphocyte arm of the immune system. These, including chronic lymphocytic leukemia and multiple myeloma, rise dramatically in incidence throughout life, with the real majority of these disorders found in elderly individuals. Whether this dramatic relationship is caused by an enhanced susceptibility of the B-lymphocyte to neoplastic transformation in older individuals is a question relevant to the entire issue of the relationship between the aging process and the neoplastic process, a subject that is considered next.
Not only does cancer occur at an increased rate in older individuals, but it makes a significant impact on such people’s lives, from the standpoint of both increasing morbidity and mortality. As Figure 94-1 also demonstrates, the age-specific cancer mortality continues to rise as a function of age, as does incidence. In support of this observation is the report from the SEER Program that 5-year survivals for most types of cancer decrease with advancing age.
Relationship of Aging and Neoplasia
It is difficult to discuss a relationship between two processes—aging (senescence) and neoplastic transformation—both of which are still incompletely understood at this time. To explore the relationship, however, we must first briefly describe the current understanding of the multistep process of carcinogenesis. The first stage of cancer development is known as initiation. In this process, chemical or physical carcinogens, or certain viruses, cause a change in the cell that predisposes it to a subsequent malignant transformation. This change appears to be an irreversible lesion in the genomic deoxyribonucleic acid (DNA) of a stem cell; the lesion may remain stable for a long period. It is not clear whether such an initiated cell can be recognized clinically, but certain disorders such as myelodysplasia or carcinoma in situ, may be a manifestation of this phenomenon.
The next stage of carcinogenesis is called promotion and involves a proliferative phase. Promoters are agents that can induce mitogenesis, or cell division, in an initiated cell. This phenomenon includes the activation of a number of growth factors and transcription factors, which promote cell proliferation, and which may arise through the events related to changes in oncogenes or tumor suppressor genes noted later. While it appears that a single initiating event is sufficient to begin the process, promotion appears to be most successful when it is repetitive. This may occur shortly after initiation or after a delay and appears to be dose-dependent as well as reversible. For this reason, researchers believe that cessation of cigarette smoking (containing both initiators and promoters) reduces the cancer incidence of former smokers when compared with those who continue to smoke.
The final stage of cancer development in this model is progression. This is actually multiphasic itself and involves the transformation of a cell from a premalignant to a malignant state, the potential clonal evolution of a subset of such cells, and the potential development of metastasis. The latter two phenomena are quite important and have led to the concept of tumor cell heterogeneity. Although we believe that tumors arise from a single “clone of cells,” tumor cells are genetically more unstable than normal cells, yielding progeny with variable proliferative and metastatic potential. Thus, not all cells within a given tumor are the same. Clinically, this may explain such diversity as variable chemosensitivity of tumor cells, the selection of resistant cells, the differential behavior of different metastatic lesions compared with the original tumor and with other metastatic lesions, and sometimes the unpredictable behavior of a particular cancer. Other factors relating to the tumor’s impact on host tissue may also play an important role at this stage. These include the ability to disrupt stromal elements such as the basement membrane and the ability to promote angiogenesis, which supports further tumor growth.
It is clear that alterations in growth regulatory gene function play a critical role in the development of neoplasia. There are two major classes of such genes—oncogenes and tumor-suppressor genes. Oncogenes, or cancer genes, were initially described as viral genes capable of transforming normal cells to malignant ones. Of the various genes, approximately 100 to 200 have been shown to be targets for oncogenic disruption of their DNA structure. Oncogene activation can result in one of two pathways: First, the decision of a cell to divide or undergo senescence and apoptosis (programmed cell death) is altered such that the balance is shifted away from cell death and toward cell survival and proliferation. Second, the oncogene mutations might affect DNA repair, thus predisposing cells to additional DNA damage and activation of additional oncogenes. Oncogenes can be either dominant or recessive. Dominant oncogenes are those for which a functional alteration in one allele contributes to the malignant phenotype despite the persistence of the normal contralateral allele. Recessive oncogenes are called tumor suppressor genes, which normally limit cell growth, enhance apoptosis, and control differentiation and require both alleles to be disrupted to contribute to the cancer process.
A large number of oncogenes have been described. They appear to have the potential for growth-enhancing activity at a series of steps along the mitogenic pathway, including activating signal transduction at the cell surface, producing endogenous growth factors at the cytoplasmic level (e.g., ras-like), and increasing the sensitivity of the cell to exogenous (or endogenous) growth factors at the nuclear level (e.g., myc-like). Another oncogene, bcl-2, codes for an inner mitochondrial protein that blocks apoptosis, or programmed cell death. This mechanism may be of particular interest in the context of senescence, as it may operate by increasing cell longevity rather than proliferation. Increased cellular oncogenic expression has been noted in many tumors and can be mimicked experimentally by altering the DNA encoding for the oncogene, usually at or near the promoter. Thus, it is possible that the chromosomal damage noted in neoplasia during the initiation and promotion phases, if it occurred near the region of an oncogene, could result in transformation and clonal evolution of the cancer. The evolutions noted in Burkitt lymphoma and chronic granulocytic leukemia may be examples of this process. The expression of more than one oncogene is necessary to cause transformation.
A number of tumor-suppressor genes have been described, and mutations in such genes as Rb and p53 have been described in many human tumors. The normal function of such genes appears to prevent uncontrolled growth as a result of the action of various growth-promoting factors. It has even been suggested that senescence may act as a form of tumor suppression. In fact, the two tumor-suppressor genes just noted, p53 and Rb, are essential for maintaining the senescent phenotype of cells in culture. Inactivation of either gene extends the replicative life span of such cells, and mutations of Rb have been linked to both cancer incidence and longevity in mice. This phenomenon would appear to enhance the likelihood of acquiring further mutations, leading to the ultimate development of malignancy. Indeed deletions of p53, Rb, or both, are frequently found in common solid tumors, such as lung, breast, and colon. Although the inactivation of tumor suppressor genes is important in the development of neoplasia, it is likely that alterations in both oncogenes and tumor-suppressor genes are necessary in many cases to achieve full malignant potential.
It is also important to understand the theory of the “two-hit hypothesis.” As described above, certain tumor suppressor genes are responsible for suppressing the malignant phenotype. In normal individuals, both alleles of such genes would need to be eliminated by random or environmentally induced errors thus requiring “two-hits.” In contrast, families with certain genetic mutations that make them susceptible to cancer already have one abnormal allele of the tumor suppressor gene. Thus, the “first-hit” preexists making them substantially more susceptible to carcinogenic action of the “second random hit.”
How then might the aging process influence the process of neoplastic transformation to result in the markedly increased rates of cancer in older people? General aspects of the aging process were discussed in Chapter 1, and only certain specific aspects relevant to the process under discussion will be reiterated here. Table 94-2 lists the types of theories that appear relevant to an explanation of the striking epidemiologic relationship.
Longer duration of carcinogenic exposure |
Increased susceptibility of cells to carcinogens |
Decreased ability to repair DNA |
Oncogene activation or amplification |
Decrease in tumor suppression gene activity |
Telomere shortening and genetic instability |
Microenvironment alterations |
Decreased immune surveillance |
Longer duration of carcinogenic exposure: It is possible that aging simply allows the time necessary for the accumulation of cellular events to develop into a clinical neoplasm. There is evidence for age-related accumulation and expression of genetic damage. Somatic mutations are believed to occur at the rate of approximately 1 in 10 cell divisions, with approximately 10 cell divisions occurring in a lifetime of a human being. Certainly, the complex set of events required in the multistep process of carcinogenesis, for example, as described for colon cancer in humans, does occur over time. The passage of time alone, however, is not likely to explain the phenomenon, as the time for a mutated cell to become a malignant cell and then subsequently to become a detectable tumor has been estimated to be approximately 10% to 30% of the maximum life span for a given animal species, which may vary from just a few years to more than 100 years.
Altered susceptibility of aging cells to carcinogens: Data in this area are somewhat contradictory. In some cases, the incidence of skin tumors in mice produced with benzopyrene has been more related to dose than to age, whereas in other models, accelerated carcinogenesis as a function of age has been demonstrated, as, for example, when dimethyl benzanthracene (DMA) was applied to skin grafts of young and old mice. In addition, an age-related increase in the sensitivity of lymphocytes to cell-cycle arrest and chromosome damage after radiation has been demonstrated. It is also possible that there are alterations in carcinogen metabolism with age, but the findings from such studies have also been contradictory.
Decreased ability to repair DNA: It is possible that damage, once initiated, is more difficult to repair in older cells. A number of studies demonstrate decreased DNA repair as a function of age following damage by carcinogens as well as radiation. Such repair failures may also be reflected in increased karyotype abnormalities in aged normal cells as well as in older patients with neoplastic disease.
Oncogene activation or amplification or decrease in tumor suppressor gene activity: These processes might be increased in the older host, resulting either in increased action or promotion or in differential clonal evolution. Although evidence is currently limited, there have been observations of increased amplification of proto-oncogenes and their products in aging fibroblasts in vitro as well as evidence for increased c-myc transcript levels in the livers of aging mice. Alternatively, such factors as genetic alterations or DNA damage could lead to inactivation of cancer-suppressor genes. Given that age-related mutations frequently appear to result in the loss of function, alterations in tumor-suppressor genes may prove to be an important mechanism.
Telomere shortening and genetic instability: The function of telomeres and the enzyme telomerase appears to be intimately involved in both the senescence and neoplasia processes. Telomeres, the terminal end of all chromosomes, shorten progressively as cells age. This functional decline begins at age 30 and continues at a loss of approximately 1% per year. This shortening appears to be causally related to controlled cell proliferation and limitation of population doubling. It is thought that each time a cell divides, 30 to 200 base pairs are lost from the end of that cell’s telomeres. Because the major function of telomeres is to protect the stability of the more internal coding sequences (i.e., allow cells to divide without losing genes), the loss of this function may lead to genetic instability, which may promote mutations in oncogenic or tumor-suppressor gene sequences. Without telomeres, chromosome ends could fuse together and degrade the cell’s genetic blueprint, making the cell malfunction, become cancerous, or even die. Telomere length is a predictor of mortality in people aged 60 or older.
The enzyme telomerase is responsible for adding back telomeric repeats to the ends of chromosomes, that is, regenerate the telomeres. This enzyme is generally not expressed in normal cells, but it is activated in malignant cells. While telomerase can reverse replicative cell senescence, the indiscriminate expression of this enzyme might increase the likelihood of tumor formation. It is also possible that p53 or Rb inactivation that occurs in senescent cells, or the inactivation of other tumor-suppressor genes, may allow the activation of the telomerase gene and promote cell immortalization and ultimately malignancy.
Microenvironment alterations: Older people accumulate senescent cells as shown by β-galactosidase staining. They also have higher levels of IL-6, which is one of the causes of frailty. A number of factors in the tumor microenvironment are critical for the development of the malignant phenotype, especially invasion and metastasis. Senescent cells can compromise tissue renewal capacity, and secrete multiple factors that alter tissue homeostasis. These factors include inflammatory cytokines like IL-1 and epithelial growth factors (e.g., heregulin, and matrix metalloproteinases like MMP-3) and can disrupt the architecture and function of the surrounding tissue and stimulate or inhibit the proliferation of neighboring cells.
Senescent fibroblasts disrupt epithelial alveolar morphogenesis, functional differentiation, and branching morphogenesis. Senescent human and mouse fibroblasts also increase vascular endothelial growth factor expression, and hypoxia further induces endothelial growth factor in senescent cells. Thus, senescent cells can create a tissue environment that synergizes with mutation accumulation to facilitate the progression of malignancies.
Decreased immune surveillance: A decrease in immune surveillance, or immunosenescence, could contribute to the increased incidence of malignancies. With respect to tumor-related immunity, however, there is a considerable amount of evidence in animal models for a loss of tumor-specific immunity with progressive age. This includes the altered capacity of old mice to reject transplanted tumors, the close relationship between susceptibility to malignant melanomas and the rate of age-related T-cell-dependent immune function decline, and the ability by immunopharmacologic manipulation to increase age-depressed tumoricidal immune function and to decrease the incidence of spontaneous tumors. The evidence linking such data to age-associated immune deficiency and the rise of cancer incidence in humans, however, is mainly circumstantial and not likely to be fully explanatory, as the types of tumors seen in the most striking examples of immune deficiency are very different from those seen in the usual aging human.
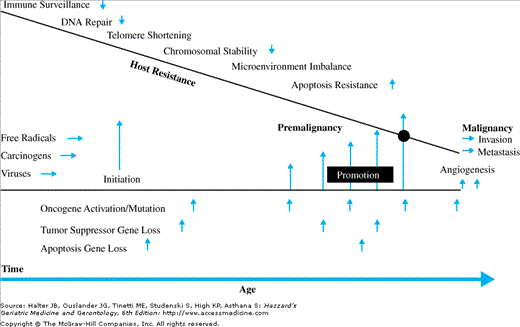
Figure 94-2 summarizes in a schematic fashion the potential interaction of these many factors that may be of importance in the increase of cancer with age. It indicates the interface of time and age-related events, such as free radical and other carcinogenic exposure, resulting in initiation, then cumulative promoting events, including mutations and other alterations in critical genes, which alternately exceed a threshold of host resistance factors, which have been progressively reduced during the aging process.
Cellular senescence suppresses cancer by arresting cells at risk of malignant transformation. However, senescent cells also secrete molecules that can stimulate premalignant cells to proliferate and form tumors, suggesting that the senescence response is antagonistically pleiotropic. Thus, cellular senescence-induced suppression of malignant transformation, a function important for the organism in early life (through the reproductive period), may be selected for, despite the fact that cellular senescence may be quite injurious in later life. In this case, senescence could be viewed as the price we pay in later life for the rigorous attempt to control proliferation to avoid neoplasia early on. The progressive, and no doubt multifactorial, loss of the controls with aging ultimately results in an increase in neoplasia in advanced age despite the early control. From the standpoint of natural selection, both of these outcomes would be acceptable, as they tend to occur long after the reproductive life span. It is likely that through research at the basic level concerning the interactions of aging and neoplasia, we will learn a great deal about the fundamental basis of each. It is hoped that such information will enhance our ability to engage in prevention at the primary and secondary levels.
A large proportion of cancers are potentially preventable. The most obviously available modalities in this regard are avoidance of known and suspected carcinogenic exposures such as tobacco smoke, occupational and environmental chemicals, excessive sunlight, and dietary factors such as excessive fat and smoked, salted, and pickled foods. Although older individuals have potentially acquired a lifetime exposure to such carcinogens, they should still accrue benefits from modifying these behaviors as well as from engaging in positive ones such as the suggested intake of fiber, vitamins, and fresh vegetables. In recent years, there has been increased interest in cancer prevention through chemopreventive intervention approaches. However, results are inconsistent and some have shown adverse effects. The evidence for tamoxifen as a preventive agent for breast cancer is the strongest, but the agent appears to be less effective in elderly women.
In 2006 the Food and Drug Administration (FDA) approved the use of a human papillomavirus-16 (HPV-16) live particle vaccine to prevent cervical cancer, precancerous genital lesions, and genital warts caused by HPV types 6, 11, 16, and 18. The vaccine is approved for use in females 9 to 26 years of age and has not been studied in elderly patients. The Selenium and Vitamin E Cancer Prevention Trial (SELECT) is an ongoing randomized, prospective, double-blind study designed to determine whether selenium and vitamin E alone and in combination can reduce the risk of prostate cancer among healthy men. The final results from this study are anticipated in the year 2013. Thus, currently there is no compelling evidence for the efficacy of the use of chemopreventive agents among the elderly population, and it still must be considered experimental.
Clinical Presentations and Disease Behavior




