PRINCIPLES OF TUMOR VIROLOGY
Viral infections are estimated to play a causal role in at least 11% of all new cancer diagnoses worldwide.
1 A vast majority of cases (>85%) occur in developing countries, where poor sanitation, high rates of cocarcinogenic factors such as HIV/AIDS, and lack of access to vaccines and cancer screening all contribute to increased rates of virally induced cancers. Even in developed countries, where effective countermeasures are widely available, cancers attributable to viral infection account for at least 4% of new cases.
2,3Viruses thought to cause various forms of human cancer come from six distinct viral families with a range of physical characteristics (
Table 5.1). All known human cancer viruses are capable of establishing durable, long-term infections and cause cancer only in a minority of persistently infected individuals. The low penetrance of cancer induction is consistent with the idea that a virus capable of establishing a durable productive infection would not benefit from inducing a disease that kills the host.
4 The slow course of cancer induction (typically over a course of many years after the initial infection) suggests that viral infection alone is rarely sufficient to cause human malignancy and that virally induced cancers arise only after additional oncogenic “hits” have had time to accumulate stochastically.
In broad terms, viruses can cause cancer through either (or both) of two broad mechanisms: direct or indirect. Direct mechanisms, in which the virus-infected cell ultimately becomes malignant, are typically driven by the effects of viral oncogene expression or through direct genotoxic effects of viral gene products. In most established examples of direct viral oncogenesis, the cancerous cell remains “addicted” to viral oncogene expression for ongoing growth and viability.
A common feature of DNA viruses that depend on host cell DNA polymerases for replication (e.g., papillomaviruses, herpesviruses, and polyomaviruses) is the expression of viral gene products that promote progression into the cell cycle. A typical mechanism of direct oncogenic effects is through the inactivation of tumor suppressor proteins, such as the guardian of the genome, p53, and retinoblastoma protein (pRB). This effectively primes the cell to express the host machinery necessary for replicating the viral DNA. The study of tumor viruses has been instrumental in uncovering the existence and function of key tumor suppressor proteins, as well as key cellular proto-oncogenes, such as Src and Myc.
In theory, viruses could cause cancer via direct hit-and-run effects. In this model, viral gene products may serve to preserve cellular viability and promote cell growth in the face of otherwise proapoptotic genetic damage during the early phases of tumor development. In principle, the precancerous cell might eventually accumulate enough additional genetic hits to allow for cell growth and survival independent of viral oncogene expression. This would allow for stochastic loss of viral nucleic acids from the nascent tumor, perhaps giving a growth advantage due to the loss of “foreign” viral antigens that might otherwise serve as targets for immune-mediated clearance of the nascent tumor. Although hit-and-run effects have been observed in animal models of virally induced cancer,
5 these effects are extremely difficult to address in humans. Currently, there are no clearly established examples of hit-and-run effects in human cancer.
In indirect oncogenic mechanisms, the cells that give rise to the malignant tumor have never been infected by the virus. Instead, the viral infection is thought to lead to cancer by attracting inflammatory immune responses that, in turn, lead to accelerated cycles of tissue damage and regeneration of noninfected cells. In some instances, virally infected cells may secrete paracrine signals that drive the proliferation of uninfected cells. At a theoretical level, it may be difficult to distinguish between indirect carcinogenesis and hit-and-run direct carcinogenesis, because, in both cases, the metastatic tumor may not contain any viral nucleic acids.
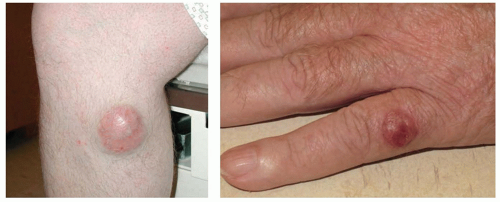
A variety of hunting approaches have been used to uncover etiologic roles for viruses in human cancer. The first clues that high-risk human papillomaviruses (HPVs), Epstein-Barr virus (EBV), Kaposi’s sarcoma-associated herpesvirus (KSHV), and Merkel cell polyomavirus (MCPyV) might be carcinogenic were based on the detection of virions, viral DNA, or viral RNA in the tumors these viruses cause. A common feature of known virally induced cancers is that they are more prevalent in immunosuppressed individuals, such as individuals suffering from HIV/AIDS or patients on immunosuppressive therapy after organ transplantation. This is thought to reflect the lack of immunologic control over the cancer-causing virus. Studies focused on AIDS-associated cancers provided the first evidence for the carcinogenic potential of KSHV and MCPyV. A theoretical limitation of this approach is that some virally induced cancers may not occur at dramatically elevated rates in all types of immunosuppressed subjects, particularly if the virus causes only a fraction of cases (e.g., HPV-induced head and neck cancers). Fortunately, the unbiased analysis of nucleic acid sequences found in tumors has become substantially more tractable as deep-sequencing methods have continued to fall in price. In the coming years, it should be increasingly possible to search for viral sequences without making the starting assumption that all virally induced tumors are associated with immunosuppression.
6One limitation of tumor sequencing approaches is that they might miss undiscovered divergent viral species within viral families known to have extensive sequence diversity
7 and could miss viral families that have not yet been discovered.
8 Tumor-sequencing approaches might also miss viruses that cause cancer by hitand-run or indirect mechanisms. It is conceivable that this caveat could be addressed by focusing on sequencing early precancerous lesions thought to ultimately give rise to metastatic cancer.
An additional successful approach to hunting cancer viruses involves showing that individuals who are infected with a particular virus have an increased long-term risk of developing particular forms of cancer. This approach was successful for identifying and validating the carcinogenic roles of high-risk HPV types, hepatitis B virus (HBV), hepatitis C virus (HCV), KSHV, and human T-lymphotropic virus 1 (HTLV-1). Although viruses that are extremely prevalent, such as EBV and MCPyV, are not amenable to this approach per se, it may still be possible to draw connections
between cancer risk and either unusually high serum antibody titers against viral antigens or unusually high viral load. Relatively high serologic titers reflect either comparatively poor control of the viral infection in at-risk individuals or expression of viral antigens in tumors or tumor precursor cells.
9,10The finding that a virus causes cancer is good news, in the sense that it can suggest possible paths to clinical intervention. These can include the development of vaccines or antiviral agents that prevent, attenuate, or eradicate the viral infection and thereby prevent cancer; the development of methods for early detection or diagnosis of cancer based on assays for viral nucleic acids or gene products; or the development of drugs or immunotherapeutics that treat cancer by targeting viral gene products. Unfortunately, establishing the carcinogenicity of a given viral species is an arduous process that must inevitably integrate multiple lines of evidence.
11 The demonstration that the virus can transform cells in culture and/or cause cancer in animal models provides circumstantial evidence of the oncogenic potential of a virus. All known human cancer viruses meet this criterion. However, it is important to recognize that viruses can theoretically coevolve to be noncarcinogenic in their native host (e.g., humans) and cause cancer only in the dysregulated environment of a nonnative host animal. This caveat may apply to human adenoviruses.
Finding that viral DNA is clonally integrated in a primary tumor and its metastatic lesions helps address the caveat that the virus might merely be a hitchhiker that finds the tumor cell a conducive environment in which to replicate (as opposed to playing a causal carcinogenic role). This caveat is also addressed by the observation that, in most instances, viruses found in tumors have lost the ability to exit viral latency and are functionally unable to produce new progeny virions. An unfortunate consequence of this is that vaccines or antiviral agents that target virion proteins (e.g., vaccines against high-risk HPVs or HBV) or gene products expressed late in the viral life cycle (e.g., herpesvirus thymidine kinase, which is the target of drugs such as ganciclovir) are rarely effective for treating existing virally induced tumors.
Demonstrating that a vaccine or antiviral agent targeting the virus either prevents or treats human cancer is by far the strongest form of evidence that a given virus causes human cancer. This type of proof has fully validated the causal role of HBV in human liver cancer. Compelling clinical trial data also show that antiherpesvirus therapeutics can prevent KSHV- or EBV-associated lymphoproliferative disorders, and that vaccination against HPV can prevent the development of precancerous lesions on the uterine cervix.