Nonopioid Analgesics
The World Health Organization recommended nonopioid analgesic agents and antipyretics as basic drugs for the management of pain, and clinicians should be familiar with the use, efficacy, and adverse effects of these agents. Numerous studies on the pharmacotherapy of cancer pain recommend the use of nonopioid analgesics for management.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 Advantages in using these drugs include their ability to control pain by an independent mechanism from alternative analgesics (eg, in the management of bone pain) or that they may help reduce the dose of opioid required for pain control (opioid-sparing effect).
A wide range of drugs with varying effects and side effects are available. Three groups of drugs are considered nonopioid analgesics: acetylsalicylic acid (ASA), acetaminophen, and nonsteroidal anti-inflammatory drugs (NSAIDs). Although ASA may be considered an NSAID, it merits separate discussion because it is effective, cheap, and widely available.
The basic mechanism of action of all NSAIDs is by inhibition of prostaglandin biosynthesis through effects on the enzyme cyclooxygenase (COX). In addition to peripheral effects, NSAIDs exert a central action at the brain or spinal cord that may influence analgesic effects.15 The formation of prostaglandins causes inflammation, swelling, pain and fever. Acetaminophen has a different mechanism of action which appears to be a combination of central COX inhibition and a central serotonergic effect.16 A homogeneous, enzymatically active COX or prostaglandin endoperoxide synthase (PGHS) was first isolated in 1976. This membrane-bound hemoprotein and glycoprotein is found in greatest amounts in the endoplasmic reticulum of prostanoid-forming cells.17 It exhibits COX activity that both cyclizes arachidonic acid and adds the 15-hydroperoxy group to form PGG2. The hydroperoxy group of PGG2 is reduced to the hydroxy group of PGH2 by a peroxidase that uses a wide variety of compounds to provide the requisite pair of electrons. Both COX and hydroperoxidase activities are contained in the same dimeric protein molecule. NSAIDs inhibit only the COX reaction of PGH synthase. Prostaglandins are important in the regulation of thrombocyte aggregation, inflammatory processes, pain and fever induction, the regulation of blood flow, and many other processes. Structural analysis has shown at least two isoforms of COX, COX-1 and COX-2 (Fig. 17.1). COX-2 is inducible and appears to have both physiologic and pathophysiologic roles (Fig. 17.2).
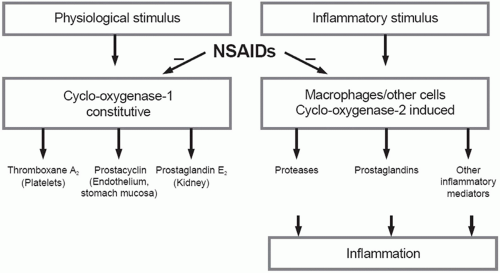 FIGURE 17.1 Regulation of prostaglandin biosynthesis by COX-1 and COX-2. (From Steinmeyer J. Pharmacological basis for the therapy of pain and inflammation with nonsteroidal anti-inflammatory drugs. Arthritis Res. 2000;2:379-385. Reprinted with permission of Biomedcentral.) |
Epidemiological studies associated a decreased incidence of colorectal cancer with the long-term use of aspirin.18 Reports also suggested that the use of NSAIDs (in particular sulindac) showed evidence of regression of colorectal adenomas.19 In subsequent years, the use of other NSAIDs, which inhibit COX enzymes, was linked to reduced cancer risk in multiple tissues including those of the breast, prostate, and lung.20 Together these studies
resulted in the identification of a new cancer preventive and/or therapeutic target-COX enzymes, especially COX-2. The overexpression of COX-2, and less consistently, the upstream and downstream enzymes of the prostaglandin synthesis pathway, was demonstrated in multiple cancer types and some preneoplastic lesions.21 Direct interactions of prostaglandins with their receptors through autocrine or paracrine pathways to enhance cellular survival or stimulate angiogenesis have been proposed as the molecular mechanisms underlying the procarcinogenic functions of COX-2.21
resulted in the identification of a new cancer preventive and/or therapeutic target-COX enzymes, especially COX-2. The overexpression of COX-2, and less consistently, the upstream and downstream enzymes of the prostaglandin synthesis pathway, was demonstrated in multiple cancer types and some preneoplastic lesions.21 Direct interactions of prostaglandins with their receptors through autocrine or paracrine pathways to enhance cellular survival or stimulate angiogenesis have been proposed as the molecular mechanisms underlying the procarcinogenic functions of COX-2.21
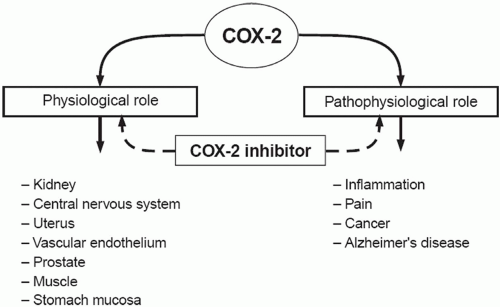 FIGURE 17.2 Physiologic and pathophysiologic functions of COX-2. (Reused with permission from Steinmeyer J. Pharmacological basis for the therapy of pain and inflammation with nonsteroidal anti-inflammatory drugs. Arthritis Res. 2000;2(5):379-385. Reprinted with permission of Biomedcentral) |
THE SALICYLATES
Salicylic or ortho-hydroxybenzoic acid belongs to a diverse group of plant phenolics (Fig. 17.3). Several plants of the Spiraea genus contain salicylic-like compounds to a significant extent. Salicin is found in the willow, poplar trees, and the black haw plant. Over 160 years ago, the chemical oxidation of salicin resulted in salicylic acid, and the acetylated derivative of this turned into one of the most successful drugs in history, aspirin. Salicylates are compounds with an aromatic ring bearing a hydroxyl group or its functional derivative. Salicylates generally act by virtue of their content of salicylic acid. Substitutions on the carboxyl or hydroxyl groups change the potency or toxicity of salicylate agents. Substitution on the benzene ring has also been accomplished. Diflunisal is a diflurophenyl derivative of salicylic acid. Salicylic acid derivatives include aspirin, sodium salicylate, choline magnesium trisalicylate, salsalate, diflunisal, salicylsalicylic acid, sulphasalazine, and olsalazine. Salicylic acid is so irritating that it can be used only externally; therefore, various derivatives of this acid have been synthesized for systemic use. These comprise two large classes, esters of salicylic acid obtained by substitution in the carboxyl group and salicylate esters of organic acids in which the carboxyl group of salicylic acid is retained and substitution is made in the hydroxyl group. Aspirin is an ester of acetic acid and there are salts of salicylic acid. Salicylate salts such as choline magnesium trisalicylate and salsalate are effective analgesics and produce fewer gastrointestinal (GI) side effects than aspirin. Aspirin selectively acetylates the hydroxyl group of one serine residue (Ser 530) located 70 amino acids from the C terminus of the enzyme.22 Acetylation leads to irreversible COX inhibition; thus, a new enzyme must be synthesized before more prostanoids are produced.
The two most commonly used preparations of salicylate for systemic effects are sodium salicylate and aspirin. A combination of choline and magnesium salicylate is also available. Mesalamine (5-ASA) is a salicylate used for its local effects in the treatment of inflammatory bowel disease. The drug is not effective orally and is usually administered as a suppository or enema. Sulfasalazine contains mesalamine linked covalently to sulfapyridine. Orally ingested salicylates are absorbed rapidly, partly from the stomach but mostly from the upper small intestine. Appreciable concentrations are found in the plasma in less than 30 minutes; a peak level is reached in about 2 hours and then gradually declines. There is little meaningful difference between the rates of absorption of sodium salicylate, aspirin, and the numerous buffered preparations of salicylates. Rectal absorption of salicylate is usually slower than oral and it is incomplete and unreliable. The plasma half-life for aspirin is approximately 15 minutes; that for salicylate is 2 to 3 hours in low doses and about 12 hours at usual anti-inflammatory doses. Aspirin absorption follows first-order kinetics with an absorption half-life ranging from 5 to 16 minutes. Hydrolysis of aspirin to salicylic acid by nonspecific esterases occurs in the liver and, to a lesser extent, the stomach, so that only 68% of the dose reaches the systemic circulation as aspirin.23 The serum half-life of salicylic acid is dose-dependent; thus, the larger the dose employed, the longer it will take to reach steady-state. Diflunisal is almost completely absorbed after oral administration, and peak concentrations occur in plasma within 2 to 3 hours. At usual analgesic doses (500 to 750 mg/day) the plasma half-life ranges between 8 and 12 hours.24
The antipyretic activity of aspirin probably results from the secondary inhibition of pyrogen-induced release of prostaglandins in the central nervous system (CNS) and possibly from centrally mediated peripheral vasodilation. Aspirin exerts its antiplatelet activity through the inhibition of COX, which is involved in the production of a platelet activator, thromboxane A2 (TXA2), and a platelet inhibitor, prostaglandin I2 (PGI2), also known as prostacyclin.25 Aspirin acetylates platelet cyclooxygenase and
prevents the synthesis of thromboxane A2, a potent vasoconstrictor and inducer of platelet aggregation.
prevents the synthesis of thromboxane A2, a potent vasoconstrictor and inducer of platelet aggregation.
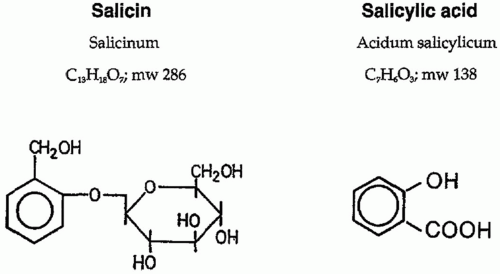 FIGURE 17.3 The molecular structure of salicin and salicylic acid. |
Low doses of aspirin reduce both pain and fever, whereas the anti-inflammatory action of aspirin requires a much higher dose. It is possible that inhibition of COX-1 is the major action of aspirin involved in its analgesic and antipyretic effects, and inhibition of COX-2 is responsible for its anti-inflammatory action.26 The standard dose of aspirin is 500 to 600 mg every 4 to 6 hours. The administration of more than 4 g/day is likely to lead to toxic effects. A review of aspirin’s effect on the renal function of experimental animals following acute and chronic administration27 revealed that although the findings are inconsistent, chronic administration can cause analgesicassociated nephropathy (AAN).
Nonacetylated salicylates, such as choline magnesium trisalicylate (CMT) and salsalate, have less ulcerproducing potential than aspirin, and at usual clinical doses do not impair platelet aggregation.28 Kilander and Dotevall29 compared the effect of aspirin and CMT on gastric and duodenal mucosa by gastroduodenoscopy after 5-day periods of administration in 10 healthy volunteers. Serum salicylate levels were similar in the two groups. All subjects given aspirin developed multiple mucosal lesions, but in only four subjects given CMT were slight mucosal changes noted, suggesting that the risk of developing mucosal lesions is much less during treatment with CMT than with aspirin. Danesh et al.30 measured parameters of platelet thromboxane biosynthesis 24 hours after ingestion of equivalent salicylate doses (500 mg) of aspirin and CMT. In random order, 10 healthy volunteers received these drugs on 2 separate days, 2 weeks apart. Whereas aspirin significantly prolonged bleeding time and decreased plasma thromboxane generation and serum thromboxane B2 levels, CMT failed to produce such effects. CMT has no inhibitory effect on platelet thromboxane biosynthesis, and may therefore be considered safer than aspirin for therapeutic use, when inhibition of platelet function can be hazardous. Because of the association with Reye’s syndrome, aspirin and other salicylates are contraindicated in children and young adults less than 12 years old with fever associated with viral illness.31
The role of salicylates is well established in inflammatory bowel disease (predominantly for ulcerative colitis and to a lesser extent in Crohn disease). Agents used for treatment include 5-aminosalicylic acid (5-ASA) or sulphasalazine (5-ASA in combination with a sulphonamide, sulphapyridine). With the aim of achieving the same efficacy of sulphasalazine but without its major side effects, various preparations of new salicylates (such as mesalazine, olsalazine, and balsalazide) have been formulated to assure targeted release, minor discomfort, and safety in use.32
We do not routinely use salicylates for the management of cancer pain, because of our concern about the toxicity associated with these agents, particularly in the immunosuppressed cancer patient, and because of their limited efficacy for pain relief. If this class of medication is deemed useful, we prefer the use of trilisate over the other agents in this group.
ACETAMINOPHEN
Acetaminophen or N-Acetyl-Para-Amino-Phenol (APAP) is one of the most widely used analgesic and antipyretic agents in the world.33 APAP, indicated for noninflammatory pain and for fever control, is available in a variety of oral preparations and as a suppository. It has potent antipyretic and mild to moderate analgesic actions but very weak anti-inflammatory activity. When administered to humans, it reduces levels of prostaglandin metabolites in urine but does not reduce synthesis of prostaglandins by blood platelets or by the stomach mucosa. Because APAP is a weak inhibitor in vitro of both COX-1 and COX-2, the possibility exists that it also inhibits COX-3,34 although Hinz et al.35 stated that APAP has substantial COX-2 inhibition in humans. APAP has been described as a preferential inhibitor of central rather than peripheral cyclooxygenases36 and is generally considered to be a weak inhibitor of the synthesis of prostaglandins. Others believe the analgesic and antipyretic benefits of APAP are due to its unique ability to selectively block COX products (principally PGE2) in the central and/or peripheral nervous system.37 The mechanism of the analgesic action also involves the serotonergic system and appears to act predominantly on descending inhibitory pathways.38 There is a central serotonergic mechanism of action that is not stimulus-dependent although its primary site of action may still be inhibition of prostaglandin synthesis.39 The use of 5-HT3 antagonists such as tropisetron and granisetron with APAP completely blocked the analgesic effect of APAP in humans.16 APAP-induced antinociception also involves the cannabinoid system.40
APAP is not considered to influence platelet function in vivo. Studies using conventional doses (approximately 1 g) of oral APAP have shown negative results.41, 42 However, Munsterhjelm et al.43 demonstrated that intravenous APAP (15, 22.5, and 30 mg/kg) inhibited platelet aggregation and TxB2 release in healthy volunteers in a dose-dependent fashion. No evidence exists for the development of chronic analgesic nephropathy with APAP alone.44 Epidemiologic studies in healthy individuals have failed to demonstrate a significant correlation between APAP use and chronic renal disease and classic analgesic nephropathy.45 The only reports on this subject suggest that combination therapy with aspirin46 or ingestion of toxic doses of APAP47 may induce nephrotoxicity.
Hepatoxicity is predominantly associated with the use of APAP, and occurs after single, acute, or short-term exposure to larger (> 10 g) doses or long-term exposure with lower (> 4 g) doses, especially in alcoholics.48, 49 APAP can cause acute liver damage when massive doses
are ingested; however, even in overdose situations, the risk of severe liver damage is less than 10% with a mortality rate less than 2%, even before availability of N-acetylcysteine treatment.50 Benson et al.51 suggest that APAP can be used safely in patients with liver disease at currently recommended doses and is preferred to aspirin and other NSAIDs because of the absence of inhibitory actions on platelet aggregation or gastrointestinal and renal toxicity.
are ingested; however, even in overdose situations, the risk of severe liver damage is less than 10% with a mortality rate less than 2%, even before availability of N-acetylcysteine treatment.50 Benson et al.51 suggest that APAP can be used safely in patients with liver disease at currently recommended doses and is preferred to aspirin and other NSAIDs because of the absence of inhibitory actions on platelet aggregation or gastrointestinal and renal toxicity.
APAP is metabolized in the liver primarily by glucuronidation and sulfation. With therapeutic doses (≤ 4 g daily), only 4% is converted by the cytochrome P450 (CYP) system into the reactive toxic intermediate N-acetyl-p-benzoquinoneimine (NAPQI).52 This toxic metabolite is rendered nontoxic by binding to glutathione. With APAP overdose, CYP enzyme induction, glutathione depletion, or the inhibition of glucuronidation, this reactive metabolite cannot be sufficiently neutralized. Instead, NAPQI reacts with the cysteine group of hepatocellular proteins, which leads to the loss of cell function and cell death. Chronic alcohol use leads to a short-term, twoto threefold increase in hepatic CYP2E1, the major cytochrome responsible for production of the toxic NAPQI metabolite of APAP.53 Although the minimal amount of APAP needed to produce severe hepatotoxicity in the setting of chronic alcohol use has not been determined, the OTC (Over-The-Counter) Advisory Committee of the FDA recommends that patients who consume three or more alcoholic beverages daily should consult their physicians before use of APAP.54
The APAP-alcohol interaction is complex with acute and chronic ethanol having opposite effects. In animals, chronic ethanol causes induction of hepatic microsomal enzymes and increases APAP hepatotoxicity as expected (ethanol primarily induces CYP2E1 and this isoform is important in the oxidative metabolism of APAP).55, 56, 57 However, in man, chronic alcohol ingestion causes only modest (about twofold) and short-lived induction of CYP2E1, and there is no corresponding increase in the toxic metabolic activation of APAP.58 The APAP-ethanol interaction is not specific for any one isoform of cytochrome P450, and it seems that isoenzymes other than CYP2E1 are primarily responsible for the oxidative metabolism of APAP in man. Acute ethanol inhibits the microsomal oxidation of APAP both in animals and man.59 This protects against liver damage in animals and there is evidence that it also does so in man.60 The protective effect disappears when ethanol is eliminated and the relative timing of ethanol and APAP intake is critical.61, 62 Because the minimum safe dose of acetaminophen is not known in the setting of chronic alcohol use, it seems prudent in such situations to avoid acetaminophen altogether, especially during brief periods of abstinence.63
Drug interactions with acetaminophen are important in oncology patients. Hylek et al.64 noted a highly significant dose-response relationship between acetaminophen and coumadin. Patients who reported taking the equivalent of at least 4 regular strength (325-mg) tablets per day for longer than 1 week had a tenfold increased risk of having an INR greater than 6.0 while also taking coumadin. The risk decreased with lower intake of acetaminophen, particularly when taking 6 or fewer 325-mg tablets per week. Parra et al.65 also noted a clinically significant interaction between coumadin and daily use of acetaminophen but at higher doses of 2 to 4 g per day. Patients with low glutathione stores, such as the elderly, may have an increased risk for drug interaction between coumadin and acetaminophen.66 In addition, the mechanism of interaction may be due to an effect of acetaminophen or one of its metabolites on factors VII and IX.67
Therapeutic dosing of acetaminophen is associated with increased risk of acute liver injury.68 Use of acetaminophen for 10 days may elevate alanine aminotransferase in moderate alcohol drinkers.69 Aminotransferase elevations were reported in healthy adults receiving 4 g of acetaminophen daily.70 Studies on acetaminopheninduced chronic renal failure have produced inconclusive results,45 although some studies suggest an association between acetaminophen use and adverse chronic renal outcomes.71, 72
Overall, there are a limited number of studies specifically investigating the role of acetaminophen in cancer pain. In a double-blind, placebo-controlled, cross-over study of 30 cancer patients, Stockler et al.73 demonstrated that the addition of acetaminophen improved pain and well-being without major side effects in patients on an opioid regimen. Patients received 1 g acetaminophen every 4 hours five times a day for 48 hours. Axelsson et al.74 recommended the addition of acetaminophen to the use of “strong” opioids for cancer-related pain. Rodriquez et al.75 reported that combination opioids (codeine or hydrocodone) with acetaminophen was safe and effective in patients with moderate or severe chronic, cancerrelated pain although 34% of patients in the codeine/acetaminophen group and 29% in the hydrocodone/acetaminophen group failed to achieve pain relief.
We occasionally use acetaminophen in the management of cancer pain. In general, this drug is well tolerated in the oncology population but its efficacy is limited. It is not unusual for us to use acetaminophen in “pain cocktail” (liquid solution of methadone and other drugs in cherry syrup) for around the clock medication administration. However, opioid and acetaminophen combinations at regular daily dosing intervals may be problematic in reaching potentially toxic levels of acetaminophen and we are careful not to exceed the recommended dosage of approximately 4 g/day.
NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
NSAIDs comprise a group of structurally diverse but similarly acting compounds that are used for relieving signs and symptoms of inflammation, especially in treatment of rheumatic diseases. NSAIDS possess analgesic, antiinflammatory and antipyretic properties and are widely prescribed for the treatment of pain and inflammation.76 The main indication for use of NSAIDs is treatment of acute and chronic musculoskeletal pain and common indications for use include chronic polyarthritis, psoriatic arthritis, ankylosing spondylitis, osteoarthritis, gout, inflammatory soft-tissue rheumatism, low back pain, postoperative and post-traumatic inflammation, thrombophlebitis and vasculitis. The discovery of the COX isoforms led to establishing their importance in many
nonarthritic or nonpain states where there is an inflammatory component to pathogenesis, including cancer, Alzheimer disease and other neurodegenerative diseases. The applications of NSAIDs and the coxibs in the prevention and treatment of these conditions as well as aspirin and other analogues in the prevention of thromboembolic diseases constitute one of the major therapeutic developments of this century. NSAIDs are recommended for the treatment of mild cancer pain and, in conjunction with opioids, for the treatment of moderate to severe pain.77 NSAIDs have also been considered effective for certain cancer pain problems, such as bony metastases, although data to support this is limited. NSAIDs may provide additive pain relief when combined with opioids, even over extended periods of time,78 although most studies investigating the effects of NSAIDs for the treatment of cancer pain are of limited duration.11 Table 17.1 lists some of the NSAIDs in clinical use.
nonarthritic or nonpain states where there is an inflammatory component to pathogenesis, including cancer, Alzheimer disease and other neurodegenerative diseases. The applications of NSAIDs and the coxibs in the prevention and treatment of these conditions as well as aspirin and other analogues in the prevention of thromboembolic diseases constitute one of the major therapeutic developments of this century. NSAIDs are recommended for the treatment of mild cancer pain and, in conjunction with opioids, for the treatment of moderate to severe pain.77 NSAIDs have also been considered effective for certain cancer pain problems, such as bony metastases, although data to support this is limited. NSAIDs may provide additive pain relief when combined with opioids, even over extended periods of time,78 although most studies investigating the effects of NSAIDs for the treatment of cancer pain are of limited duration.11 Table 17.1 lists some of the NSAIDs in clinical use.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


