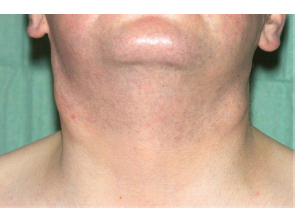
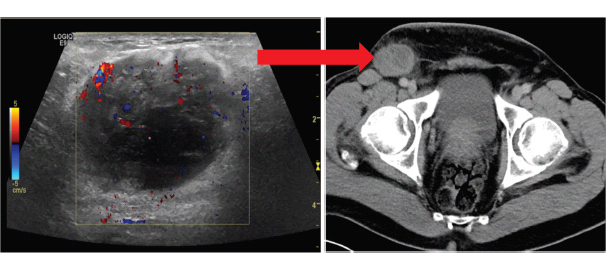
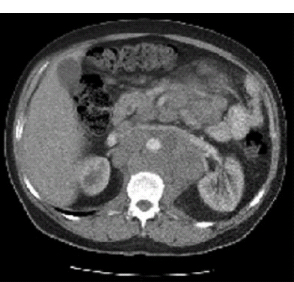
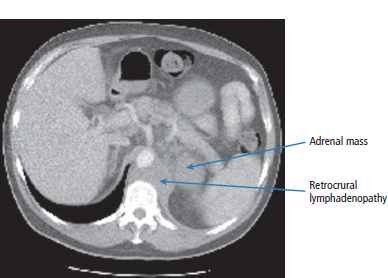
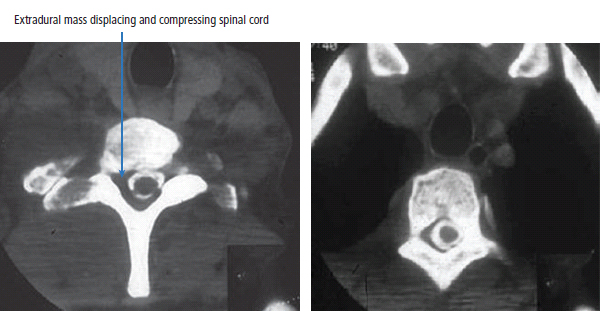
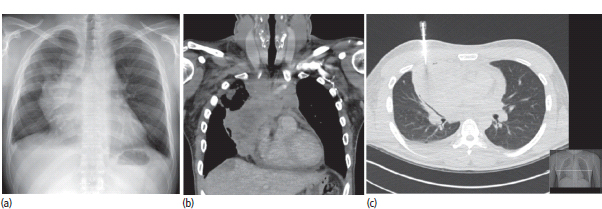
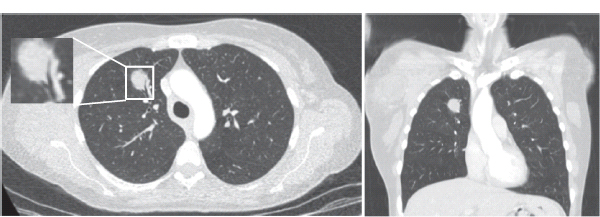
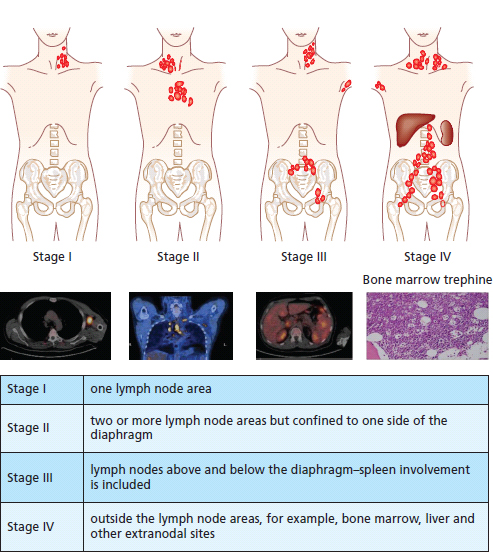
30 Non-Hodgkin’s lymphoma (NHL) is relatively common (Table 30.1). In the United Kingdom in 2011 there were 12,783 diagnoses and 4646 deaths attributable to NHL. There have been many descriptions of the pathological classification of this disease. Rather than achieving clarity, however, most have tended to confuse the situation further because of their complexity. The current WHO classification lists over 80 different forms of lymphomas! They divide the lymphomas into four groups: In terms of clinical practice, the most significant divisions are into high- and low-grade lymphoma (Table 30.2). High-grade lymphoma is 3–4 times more common than low-grade lymphoma. About 4000 people are diagnosed in the United Kingdom with low-grade lymphoma each year. Slightly more men are affected than women. Lymphomas arise from lymphoid organs or lymphatic tissue associated with other systems that contain lymphatic tissue. The latter, the so-called extranodal lymphomas, constitute up to 30% of all NHL. There have been extraordinary advances in the molecular biology of lymphoma, and from this we have begun to understand some of the aetiological features involved in this condition. The most prominent factors in the pathogenesis are infections and immune suppression. Viral infection with HIV, EBV, HHV8, HCV and HTLV are all associated with NHL (Table 30.3). Helicobacter infection in the stomach leads to a proliferation of gastric lymphoid tissue and the development of low-grade, mucosa-associated tumours. Such tumours may respond to H. pylori eradication treatment, but unfortunately they may still evolve into classical lymphoma despite eradication. In addition to infection, immunosuppression increases the risk of lymphoma including HIV infection which increases the risk of NHL by 60 times. By comparison the risk of lung cancer is increased 17 times in smokers. Similarly allograft transplant recipients, who are prescribed immunosuppressants to prevent graft rejection, are also at greatly increased risk of lymphoma. Various autoimmune diseases also raise the risk of lymphoma: Table 30.1 UK registrations for non-Hodgkin lymphoma cancer 2010 Patients present with nodal enlargement which may be accompanied by constitutional symptoms including weight loss, sweating and fever (Figures 30.1 and 30.2). These symptoms – where weight loss is in excess of 10% of pre-morbid weight, sweating is sufficient to drench night clothes and fever exceeds 38°C – are described as “B” symptoms. “B” symptoms are less common in high-grade lymphoma than low-grade malignancies. Patients with such symptoms should be referred to specialist centres where the chance for survival and the quality of survival are significantly better than in peripheral non-specialist centres. The care of patients with lymphoma should be by oncologists or haematologists, depending upon the specialist interests of the clinicians. Table 30.2 The most common pathological subtypes of non-Hodgkin lymphoma Table 30.3 Infections associated with lymphoma African (endemic) Burkitt lymphoma Primary cerebral lymphoma Hodgkin’s lymphoma Diffuse large B-cell lymphoma Post-transplant lymphoproliferative disease Nasal T/NK non-Hodgkin lymphoma Primary effusion lymphoma (with HHV8) In outpatients, a careful history is obtained from the patient who is then examined. The investigations organized should include a blood count, renal and hepatic function tests, chest X-ray, bone marrow aspiration and trephine, and PET–CT scan of body (Figures 30.3, 30.4, 30.5, 30.6 and 30.7). These investigations are done in order to define the extent of the disease. From these investigations the clinical staging is obtained (Figure 30.8). This is defined as follows: Figure 30.1 Bilateral cervical lymphadenopathy in a man with stage IV follicular lymphoma. Figure 30.2 Right inguinal lymph node with vascular rim on colour Doppler ultrasound. Core biopsy of lymph node confirmed a diagnosis of diffuse large B-cell non-Hodgkin’s lymphoma. Figure 30.3 CT scan showing massive para-aortic lymphadenopathy to diffuse large B-cell non-Hodgkin’s lymphoma. Figure 30.4 CT scan showing extensive left retrocrural adenopathy and a left adrenal mass due to high-grade B-cell non-Hodgkin’s lymphoma. Figure 30.5 Spinal cord compression at T1 by an extradural mass of high-grade non-Hodgkin’s lymphoma. Figure 30.6 Mediastinal mass. A 35-year-old man was presented with a large anterior mediastinal mass (a, b). A diagnosis of non-Hodgkin’s lymphoma (mediastinal large B-cell) was established by a CT guided biopsy (c). He was treated with six cycles of R-CHOP and remains in remission 2 years later. Figure 30.7 Lung opacity with air bronchogram. A branching linear lucency within an opacity in the lung is known as an “air bronchogram”, seen most often with consolidation and pulmonary oedema. It is rare in primary lung cancer but is seen in lymphoma of the lung (as here). Figure 30.8 Lymphoma staging. A = without symptoms; B = with symptoms including unexplained weight loss (10% in 6 months prior to diagnosis), unexplained fever and drenching night sweats. Preliminary investigations having been organized, the patient should then proceed to a lymph node biopsy. Lymph node biopsies used to be required to describe the architectural arrangement of the tumour. In modern times, they are no longer always considered to be necessary. Sufficient material can often be obtained from core needle biopsies to define the pathological diagnosis. There are many classification systems for NHL, which include the WHO classification (Table 30.4), the Kiel classification, the Working Formulation and the Revised European and American Lymphoma Classification (REAL). For the purposes of defining treatment, the most practical classification, however, is to describe the tumour as being low or high grade. A low-grade tumour tends to have a follicular nature and to contain relatively inactive cells. A high-grade tumour contains cells that have a high index of mitotic activity, meaning that many of the cells are actively proliferating, and there is no follicular structure to the lymph node. There are variant lymphomas, such as mantle cell and Burkitt’s lymphomas, which are clinical entities with poor prognosis. Many modern techniques have been applied to the pathological diagnosis of lymphoma. Immunophenotyping using monoclonal antibodies is the most helpful, firstly, in initially distinguishing between a lymphoma or a carcinoma by using antibodies to the leukocyte common antigen (CD45), and secondly, in defining the lymphoma by using antibodies that are specific for B or T lymphocytes, such as CD20 or CD4, CD2 and CD3. T-cell receptor and immunoglobulin gene rearrangement studies are also carried out, and are helpful in describing tumour clonality. Fluorescent in situ hybridization (FISH) is also useful. This is because the observed cytogenetic abnormalities are relatively specific for subtypes NHL. Some of these are outlined in Table 30.5. Low-grade tumours are generally disseminated at diagnosis. If they are localized, that is stage I, small bulk, peripheral and without B symptoms, the treatment should be radiotherapy. For stages II–IV disease, treatment is chemotherapy with oral alkylating agents such as chlorambucil or with an intravenous chemotherapy programme known as CVP which uses cyclophosphamide, vincristine and prednisone. Table 30.4 World Health Organization (WHO) classification of lymphomas Table 30.5 Recurrent chromosomal translocations in non-Hodgkin’s lymphoma subtypes, resulting in oncogene dysregulation ALK-NPM, 50% Others, 15% Ig, immunoglobulin; MALT, mucosa-associated lymphoid tissue. *BCL2 (30%) and c-MYC (10%) rearrangements are also seen in diffuse large B-cell non-Hodgkin’s lymphoma. Chlorambucil has very little early toxicity but at high total dosages causes sterility, secondary myelodysplasia (MDS) and acute myeloid leukaemia (AML). CVP leads to hair loss, but apart from this it is without significant morbidity. Both regimens may be associated with marrow toxicity which results in admissions with neutropenic sepsis or with thrombocytopenic bleeding. The monoclonal antibody rituximab is frequently added to these cytotoxic agents. Rituximab is directed against CD20 and usually has very little toxicity apart from the possibility of a hypersensitivity reaction. Patients with stage I NHL have a 70–95% chance of cure with radiotherapy. The patient with disseminated low-grade lymphoma is not cured by treatment. Although 85% of patients achieve a complete response to therapy, this response is transient. After a median period of 18 months, the patient relapses and requires re-treatment. The average patient has four such episodes of response and relapse. Finally after a median period of 7.5 years, there is transformation to high-grade lymphoma. Because treatment for disseminated low-grade lymphoma is not curative, there have been trials of the benefit of more intensive therapy administered with curative intent in this condition. A 2012 Cochrane meta-analysis unearthed five randomized controlled trials of high-dose chemotherapy and autologous stem cell rescue in patients with low-grade follicular NHL. The meta-analysis found a “strong” benefit for this approach in terms of progression-free survival. Paradoxically, high-grade lymphomas are more likely to be confined to one lymph node group than low-grade tumours and are curable. Stage I disease may be treated with abbreviated chemotherapy (usually three cycles of R-CHOP) followed by radiotherapy. Patients with small bulk stage I NHL have a 95% chance of cure. Treatment for those with more advanced disease is with the R-CHOP regimen, and there is little evidence that more complex regimens add to the chance of cure. Seventy to eighty per cent of all patients enter remission, which is sustained in about 40–60% of cases. CHOP consists of cyclophosphamide, doxorubicin (hydroxydaunorubicin), vincristine (oncovin) and prednisolone and was introduced in the 1970s. It has remained the gold standard therapy for many high-grade lymphomas ever since with the addition of rituximab for CD20-expressing lymphomas in the late 1990s. In 1993 a pivotal trial compared CHOP with several newer chemotherapy regimens with less memorable but more exotic names (e.g. m-BACOD, ProMACE-CytaBOM and MACOP-B); CHOP emerged as the regimen with the least toxicity but similar efficacy. The addition of rituximab has resulted in RCHOP becoming the gold standard for diffuse large B-cell lymphoma (DLBL). Some higher grade lymphomas such as Burkitt lymphoma require more aggressive alternating combination chemotherapy regimens. Patients with poor-prognosis lymphomas at presentation or with recurrent high-grade lymphomas may be considered for high-dose chemotherapy with autologous or allogeneic haematogenous stem cell support. These cells are usually harvested by leucophoresis following colony stimulating factor administration, or from umbilical cord blood from neonates. Stem cell transplantation programmes may be linked with attempts to purge the stem cells of specific cell populations. Immunosuppression is required for patients receiving allografts and depends on the closeness of the HLA matching of the donor and host. HLA are the human leukocyte antigen genes of the major histocompatibility complex (MHC). Prognosis depends on a number of risk factors. There is an associated mortality rate to these procedures, which is as little as 1–2% for autologous transplants and exceeds 10% for matched unrelated donor allografting. Nevertheless, it is unlikely that ever higher doses of cytotoxic chemotherapy will improve outcomes particularly as any marginal benefit is likely to be more than offset by greater treatment-related mortality. The future is more likely to lie with scientific advances leading to rational drug design and novel therapies. For example, there are three new promising approaches to mantle cell lymphoma: bortezomib, a proteosome inhibitor; lenalidomide, a thalidomide analogue that acts as an immunomodulator; and idelalisib, a phosphoinoside-3 (PI3) kinase inhibitor. Case Study: The Eritrean woman with a swollen jaw.
Non-Hodgkin’s lymphoma
Epidemiology
Pathogenesis
Percentage of all cancer registration
Rank of registration
Lifetime risk of cancer
Change in ASR(2000–2010)
5–year overall survival
Female
Male
Female
Male
Female
Male
Female
Male
Female
Male
NHL
4
4
7th
5th
1 in 61
1 in 51
+15%
+14%
66%
62%
Presentation
Pathological subtype
Grade
Percentage of NHL diagnoses
Diffuse large B-cell lymphoma (DLBL)
High grade
31
Follicular lymphoma (FL)
Low grade
22
Marginal zone lymphoma (MZ)
Low grade
8
T-cell lymphoma (TCL)
Low or high grade
7
Small lymphocytic lymphoma
Low or high grade
6
Mantle cell lymphoma (MCL)
High grade
6
Burkitt lymphoma (BL)
High grade
3
Infection
Lymphoma subtype
Virus
Epstein barr virus (EBV)
Human herpesvirus8 (HHV8)
Primary effusion lymphoma (with EBV)
Hepatitis C virus
Splenic lymphoma with villous lymphocytes
Human T-lymphotropic virus, type I (HTLV-I)
Adult T-cell leukemia-lymphoma
Bacteria
Helicobacter pylori
Gastric mucosa associated lymphoma (MALT)
Campylobacter jejuni
Immunoproliferative small intestinal disease (IPSID) – a variant of extranodal marginal zone lymphoma
Staging and grading
Treatment
Low-grade non-Hodgkin’s lymphoma
Mature B-cell neoplasms
Chronic lymphocytic leukemia/Small lymphocytic lymphoma
B-cell prolymphocytic leukemia
Lymphoplasmacytic lymphoma (such as Waldenström macroglobulinemia)
Splenic marginal zone lymphoma
Plasma cell neoplasms
Plasma cell myeloma
Plasmacytoma
Monoclonal immunoglobulin deposition diseases
Heavy chain diseases
Extranodal marginal zone B-cell lymphoma, also called MALT lymphoma
Nodal marginal zone B-cell lymphoma
Follicular lymphoma
Mantle cell lymphoma
Diffuse large B-cell lymphoma
Mediastinal (thymic) large B-cell lymphoma
Intravascular large B-cell lymphoma
Primary effusion lymphoma
Burkitt lymphoma/leukemia
Mature T-cell and natural killer (NK) cell neoplasms
T-cell prolymphocytic leukemia
T-cell large granular lymphocytic leukemia
Aggressive NK cell leukemia
Adult T-cell leukemia/lymphoma
Extranodal NK/T-cell lymphoma, nasal type
Enteropathy-type T-cell lymphoma
Hepatosplenic T-cell lymphoma
Blastic NK cell lymphoma
Mycosis fungoides/Sézary syndrome
Primary cutaneous CD30-positive T-cell lymphoproliferative disorders
Primary cutaneous anaplastic large cell lymphoma
Lymphomatoid papulosis
Angioimmunoblastic T-cell lymphoma
Peripheral T-cell lymphoma, unspecified
Anaplastic large cell lymphoma
Hodgkin Lymphoma
Classical Hodgkin lymphomas
Nodular sclerosis
Mixed cellularity
Lymphocyte-rich
Lymphocyte depleted or not depleted
Nodular lymphocyte-predominant Hodgkin lymphoma
Immunodeficiency-associated lymphoproliferative disorders
Associated with a primary immune disorder
Associated with the HIV
Post-transplant
Associated with methotrexate therapy
Primary central nervous system lymphoma in immuno-compromised patients
Histology
Translocation
Alteration of gene function
Mechanism/features of translocation
Frequency
Follicular lymphoma
t(14;18)(q32;21)
Upregulation of BCL2 (inhibitor of apoptosis)
BCL2 relocates to IgH locus. Error in physiological IgH rearrangement. Seen rarely in normal B-cells
80%
Burkitt’s lymphoma
t(8;14)(q24;q32); t(2;8)(p12;q24); t(8;22)(q24;q11)
Upregulation of c-MYC (transcription factor for cell cycle progression/proliferation)
c-MYC relocates to IgH locus or to one of the light chain gene loci (IgL or IgK)
100%
Mantle cell lymphoma
t(11;14)(q13;q32)
Upregulation of cyclin D1 (G1 cyclin)
Cyclin D1 relocates to IgH
>90%
Diffuse large B-cell lymphoma*
t(3;14)(q27;32) and several others involving 3q27
Deregulation of BCL6 (zinc finger transcription factor)
BCL6 relocates to IgH, IgL, IgK or one of many other non-Ig loci
30–40%
Extranodal marginal zone lymphoma (MALT)
t(11;18)(q21;q21)
Gene fusion of AP12 and MLT/MALT1 genes (AP12 is inhibitor of apoptosis)
Gene fusion
20–35%
Extranodal marginal zone lymphoma (MALT)
t(1;14)(q22;q32)
Deregulation of BCL10 (apoptosis regulatory protein)
BCL10 relocates to IgH locus
<5%
Lymphoplasmacytic lymphoma
t(9;14)(q13;q32)
Deregulation of PAX5 (paired homeobox transcription factor)
PAX5 relocates to IgH locus
50%
Anaplastic large cell lymphoma
t(2;5)(p23;q35) and others involving 2p23
Gene fusion of ALK (anaplastic lymphoma kinase, a receptor tyrosine kinase) and NPM (located at 5q35) or other gene malignant transforming capacity in vitro and in vivo
Gene fusion
High-grade non-Hodgkin’s lymphoma
High-dose therapy
 ONLINE RESOURCE
ONLINE RESOURCE
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


