Non-Hodgkin’s and Hodgkin’s Lymphomas and Myeloma: Introduction
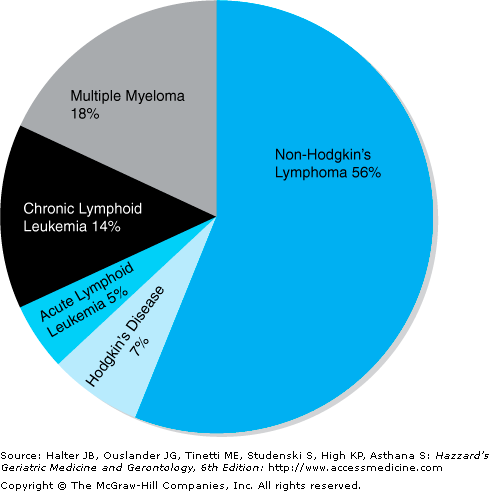
Lymphoid malignancies include nearly 40 named entities; however, they can be divided into roughly five large categories based on the clinical syndrome they cause: acute lymphoid leukemias, chronic lymphoid leukemias, non-Hodgkin’s lymphomas, Hodgkin’s disease, and plasma cell disorders (chiefly multiple myeloma). In 2007, 111,820 people were diagnosed with a lymphoid malignancy and 36,440 patients died from a lymphoid malignancy. Figure 104-1 shows a distribution of the annual incidence in a pie chart. Acute and chronic lymphoid leukemias are covered in a separate chapter. We shall discuss non-Hodgkin’s lymphomas, Hodgkin’s disease, and plasma cell disorders in this chapter.
Non-Hodgkin’s Lymphoma
The non-Hodgkin’s lymphomas (NHLs), the most common lymphoid malignancies, are a heterogeneous group of cancers that have in common the clonal expansion of cells of lymphoid origin. The heterogeneity stems from the very large number of distinct lymphocyte subsets and diverse molecular and genetic pathways to neoplasia. Mutations, chromosome translocations, or other alterations in certain genes (e.g., BCL2, c-MYC, FAS, BCL6) contribute to the pathogenesis in many cases and gene expression profiling has identified subsets of NHL with varying aggressiveness and response to chemotherapy. About 88% of all NHLs are derived from B cells. Despite insights into the alterations associated with specific NHL types, the mainstay of current therapy remains empiric and typically includes cytotoxic chemotherapy combined with monoclonal antibody directed at the CD20 molecule, which is expressed on nearly all B cells. Clinical trials suggest that elderly patients may safely receive these agents with expectations similar to their younger counterparts.
In 2007, 63,190 new cases of NHL were diagnosed and about half occurred in persons aged 60 years or older. National Cancer Institute data indicates an approximate 25% increase in NHL incidence since 1950, although there is some evidence that the rate of rise has declined somewhat since 1990. Although some of the increase can be related to the acquired immunodeficiency syndrome (AIDS) epidemic, particularly in young and middle-aged persons, the bulk of the cause of the increase is undefined, particularly in the aged population. With more effective human immunodeficiency virus (HIV) treatment, HIV-associated lymphoma is becoming less frequent. However, NHL affects older patients (one third are 70 years or older) and is the fifth leading cause of cancer deaths in women aged 80 years and older. Furthermore, current cancer surveillance statistics indicate that despite the fact that incidence is declining for the population as a whole, for those aged older than 65 years, there is a net increase of 1.3% compared to a rate of −1.5% for the younger population.
Lymphoma classification was a contentious field until 1999 when the World Health Organization Classification was developed by an international panel of hematopathologists and clinicians based on consensus criteria including histology, immunology, genetics, and clinical features of diseases (Table 104-1). The classification includes nearly 40 named entities; however, the 10 most frequent diagnoses are printed in bold type. Fortunately, about 75% of patients have one of two histological types, diffuse large B-cell lymphoma (DLBL) (~40% to 45% of lymphomas in the United States) and follicular lymphoma (FL) (~30% of lymphomas in the United States). These two prototypical forms of lymphoma highlight an intriguing paradox. In general, FLs, which have a more indolent natural history, frequently present with few symptoms but with widespread disease and are generally considered incurable with the current modalities. The more aggressive DLBLs present with more constitutional symptoms and more often at earlier stages and are in most cases curable, even at an advanced stage. The challenge for those treating lymphomas in elderly patients is to balance the likelihood of a meaningful treatment response against the potentially enhanced rate and severity of treatment toxicity in the setting of an individualized assessment of the patient’s functional status and projected longevity.
B CELL | T CELL | HODGKIN’S DISEASE |
|---|---|---|
Precursor B cell neoplasm | Precursor T cell neoplasm | Nodular lymphocyte-predominant Hodgkin’s disease |
Precursor B lymphoblastic leukemia/lymphoma (precursor B cell acute lymphoblastic leukemia) | Precursor T cell lymphoblastic lymphoma/leukemia (precursor T cell acute lymphoblastic leukemia) | |
Mature (peripheral) B cell neoplasms | Mature (peripheral) T cell neoplasms | Classic Hodgkin’s disease |
B cell chronic lymphocytic leukemia/small lymphocytic lymphoma | T cell prolymphocytic leukemia | Nodular sclerosis Hodgkin’s disease |
B cell prolymphocytic leukemia | T cell granular lymphocytic leukemia | Lymphocyte-rich classic |
Hodgkin’s disease | ||
Lymphoplasmacytic lymphoma | Aggressive NK cell leukemia | Mixed-cellularity Hodgkin’s disease |
Splenic marginal zone B cell lymphoma (± villous lymphocytes) | Adult T cell lymphoma/leukemia (HTLV-I +) | Lymphocyte-depletion Hodgkin’s disease |
Hairy cell leukemia | Extranodal NK/T cell lymphoma, nasal type | |
Plasma cell myeloma/plasmacytoma | Enteropathy-type T cell lymphoma | |
Extranodal marginal zone | Hepatosplenic γδ T cell lymphoma | |
B cell lymphoma of MALT type | ||
Mantle cell lymphoma | Subcutaneous panniculitis-like T cell lymphoma | |
Follicular lymphoma | Mycosis fungoides/Sézary syndrome | |
Nodal marginal zone B cell lymphoma (± monocytoid B cells) | Anaplastic large cell lymphoma, primary cutaneous type | |
Diffuse large B cell lymphoma | Peripheral T cell lymphoma, not otherwise specified (NOS) | |
Burkitt’s lymphoma/Burkitt cell leukemia | Angioimmunoblastic T cell lymphoma | |
Anaplastic large cell lymphoma, primary systemic type |
Given that most lymphomas are either follicular or DLBL, most of the discussion will deal with management of these two entities. Less detailed discussion will be provided for a handful of more unusual lymphomas. The approach to the patient with lymphoma is similar regardless of the specific diagnosis. One first seeks to establish the extent of the disease and establish the clinical stage (see later). A variety of specialized tools have been applied to the diagnosis of lymphomas including cytogenetics and flow cytometry to define the expression of certain cell surface markers, and immunohistochemistry and molecular biology techniques. These tests can sometimes be helpful in making a difficult diagnosis, but often the results are not available in a clinically useful time frame. Similarly, the development of microarray technology and the demonstration that various patterns of gene expression correlate with clinical outcomes is of interest; however, it is not clear that these techniques are suitable for clinical application to a particular patient.
For most lymphomas, the cause is unknown. Lymphomas are increased in the setting of primary and secondary immunodeficiency disease and certain autoimmune diseases (Sjogren’s syndrome, celiac disease, rheumatoid arthritis, systemic lupus erythematosus). A number of lymphoma types are associated with infectious agents. For example, gastric mucosa-associated lymphatic tissue (MALT) lymphoma is associated with Helicobacter pylori infection. Exposure to agricultural chemicals seems to be associated with an increased risk of lymphoma. However, the factors that produced an increasing incidence between 1950 and 1990, which has leveled off in recent years, is unexplained. Many types of lymphomas have characteristic chromosomal translocations that are thought to play a role in their origin. However, lymphomagenesis remains largely undefined. Only the c-myc translocations in Burkitt’s lymphoma are sufficient to produce the lymphoma they are associated with. Other characteristic translocations seem to be one of several molecular alterations required to produce disease.
The Ann Arbor staging system that was developed for Hodgkin’s disease is also applied to patients with NHL, but often the system applies to NHL patients poorly. With the widespread use of systemic therapy to treat all stages of NHL and Hodgkin’s disease (see later), precise pathological staging (Table 104-2) is less critical than it was when eligible patients were treated with localized therapies (e.g., radiation therapy). For example, in the low-grade lymphomas, the disease may be stage IV without much change in prognosis compared with earlier stages, and in the higher-grade aggressive lymphomas, systemic treatment is necessary even when disease is confined to one anatomical region. A patient with stage II DLBL could have small-volume disease involving two lymph node groups and an outstanding prognosis with a shortened course of chemotherapy or could instead have a football size mass in the abdomen with a poorer prognosis. Because anatomical staging does not define prognosis well in NHL, other supplementary systems have been developed. The International Prognostic Index (IPI) was developed for DLBL based on outcome of treating patients with CHOP (cyclophosphamide, hydroxydaunarubicin [doxorubicin], vincristine, prednisone) or CHOP-like regimens. The addition of rituximab to CHOP improved prognosis substantially and altered the prognostic significance of the IPI factors; these alterations have been incorporated into a new revised IPI (Table 104-3). A similar prognostic index has been developed for FL in which hemoglobin level and number of sites of nodal involvement replace Ann Arbor stage III or IV and > 1 extranodal site of disease in the IPI (Table 104-4). The staging workup for lymphomas (NHL and Hodgkin’s) is shown in Table 104-5. Aggressive histology lymphomas, such as diffuse large B cell, Burkitt’s and lymphoblastic, may require evaluation of the central nervous system (CNS) with a head computed tomogram (CT) and lumbar puncture, especially in patients with diffuse histology, bulky abdominal lymphadenopathy, or bone or bone marrow involvement. Keep in mind that older patients are more likely to present with more advanced disease. This may reflect a delay in diagnosis, as has been demonstrated for older people with solid tumors.
I | Involvement of a single lymph node region or lymphoid structure (e.g., spleen, thymus, Waldeyer’s ring) |
II | Involvement of two or more lymph node regions on the same side of the diaphragm (the mediastinum is a single site; hilar lymph nodes should be considered “lateralized” and, when involved on both sides, constitute stage II disease) |
III | Involvement of lymph node regions or lymphoid structures on both sides of the diaphragm |
III1 | Subdiaphragmatic involvement limited to spleen, splenic hilar nodes, celiac nodes, or portal nodes |
III2 | Subdiaphragmatic involvement includes paraaortic, iliac or mesenteric nodes plus structures in III1 |
IV | Involvement of extranodal sites(s) beyond that designated as “E”; more than one extranodal deposit at any location; any involvement of liver or bone marrow |
A | No symptoms |
B | Unexplained weight loss of >10% of body weight during the 6 months before staging Unexplained, persistent, or recurrent fever (T>38 degrees C) in the previous month Recurrent drenching night sweats in the previous month |
E | Localized, solitary involvement of extralymphatic tissue, excluding liver and bone marrow |
Five clinical prognostic factors:
|
Five clinical prognostic factors:
|
History, with special attention to “B” symptoms, including fevers, night sweats, and >10% loss in body weight |
Physical examination with special attention to node-bearing areas, liver, and spleen |
Initial blood work to include a complete blood count with white cell differential and platelet count, serum chemistries including liver function tests, creatinine, albumin, total protein, and erythrocyte sedimentation rate |
Radiologic studies to include chest x-ray, positron emission tomography (PET), superimposed upon chest, abdomen and pelvis CT |
Bilateral iliac crest bone marrow biopsies |
(Selected cases) Bipedal lymhangiogram |
(Selected cases) Laparotomy with exploration of lymph node–bearing areas, liver biopsies, and splenectomy; if laparotomy is not to be performed, consider percutaneous or laparoscopic-guided liver biopsies |
Overall, it does not appear that lymphomas in older patients have distinct clinical or pathological features from those same lymphomas occurring in younger people, though the confidence in that conclusion is undermined by the general underrepresentation of older patients on clinical trials. However, in certain series, it has been reported that older patients are more likely to have diffuse histology and present with extranodal disease involving gastric or bone marrow sites.
DLBL is the most common form of lymphoma in the United States accounting for 40% to 45% of cases. DLBL often presents with diffuse adenopathy or an abdominal mass, and extranodal sites are involved in about 45% of cases. Factors influencing prognosis have been incorporated into a prognostic index (Table 104-3). The median age is about 64 years. The male-to-female ratio is 55:45. About one third of patients have B symptoms and about 50% present with advanced stage disease. A small subset of patients has disease restricted largely to the mediastinum and the tumor is encased in dense fibrotic tissue. This mediastinal DLBL predominantly affects younger patients (median age 37 years) and women are affected twice as commonly as men. The approach to the patient is not influenced by the differences in presentation.
Most DLBL occurs de novo in the absence of known underlying disease. However, DLBL can also occur in the setting of immunosuppression, either from primary immunodeficiency diseases or AIDS or from iatrogenic immunosuppression in the setting of organ transplant. Often the Epstein–Barr virus (EBV) is an etiological agent of lymphomas occurring in immunosuppressed patients. EBV is also associated with primary CNS lymphoma and some cases of Hodgkin’s disease, particularly cases of mixed cellularity histology. DLBL frequently manifest mutations in p53 and alterations in the Bcl-6 gene on chromosome 3q27. Distinct subsets have been defined by gene expression profiling, but such data are not usually relevant to the management of patients.
The treatment of choice for DLBL is a five-drug regimen called CHOP-R (cyclophosphamide, doxorubicin, vincristine, prednisone and rituximab). Patients with early stage disease may receive four to six cycles and those with advanced disease six to eight cycles. Overall survival at 4 years ranges from 94% for those with no poor prognostic factors to 55% for those with three or more poor prognostic factors. Patients who do not relapse within the first 2 years of achieving a complete response are very unlikely to do so. Overall, complete response rates are 70% to 100% with less than one third of complete responders relapsing. Untreated patients survive only 4 to 6 months. About 40% of patients who relapse are curable with high-dose therapy and autologous hematopoietic stem cell transplantation. Overall, more than two thirds of patients with DLBL are cured.
The curability of the disease makes it essential that the treating physician carefully inform the patient of the options available to them. CHOP-R is generally well-tolerated with predictable dose-related toxicities. Even very old patients can be successfully treated. In addition, infusional regimens like R-EPOCH (rituximab, etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin) are also safe and highly effective in older patients.
The new standard treatment represents a true advance in therapy. In an earlier study, the Nebraska Lymphoma Study Group used the CAP/BOP regimen (cyclophosphamide, doxorubicin, procarbazine, bleomycin, vincristine, and prednisone) to treat all patients with DLBL. They found a similar overall complete remission (CR) rate of 65% in patients older and younger than 60 years of age; however, patients younger than 60 years had a 62% 5-year survival, whereas those older than 60 years had a 35% 5-year survival. They found that treatment toxicity was similar and that deaths from apparently unrelated causes accounted for most of the survival difference. However, as therapy has improved, the contribution of age to prognosis has decreased.
FL is the second most common form of lymphoma in the United States accounting for about 33% of cases. Thus, FL and DLBL together account for about 75% of lymphomas. FL is predominantly a disease of lymph nodes. New painless adenopathy is the predominant presenting symptom. Some patients give a history of adenopathy that has waxed and waned in the past. B symptoms occur in less than 25% of patients and the disease is often widely disseminated at presentation with involvement of bone marrow noted in over 40%. Females are affected slightly more commonly than males and the median age is 59 years.
FL is associated with a characteristic genetic lesion, a t(14;18) that results in the expression of bcl-2, an antiapoptotic protein. How bcl-2 expression is related to lymphomagenesis is undefined. An important biologic feature of FL is its genetic instability. Normal follicle center B cells undergo extensive somatic hypermutation to increase the ability of the antibody it produces to bind to antigens. In neoplastic follicular center cells, the mutation process is not restricted to the immunoglobulin genes. As mutations in other genes accumulate, the disease has a tendency to undergo transformation to DLBL with an acceleration in natural history and often an increased resistance to therapy. Histologic progression occurs at a rate of 5% to 7% per year and is not increased by therapy. The vast majority of patients with FL who die have DLBL as the cause of death. Median survival appears to have been increasing in the last 20 years from a median of 8 years to a median of 13 years. The basis for the improvement is not defined.
Patients in whom FL is restricted to lymph nodes may be cured by involved-field radiation therapy. Early studies of radiation therapy in early stage disease documented long-term disease-free survival in 90% of patients. The relegation of a patient to a watch-and-wait approach without staging the disease is indefensible. Many patients with localized disease have experienced disease progression while under “observation” and are thereby denied the potential for cure of early stage disease.
Management of patients with advanced stage disease is controversial. Because of the indolent course of disease, an initial period of “watchful waiting” is very appealing, especially in the elderly. In prospective trials, no overall survival difference has been demonstrated between observation and early chemotherapy. However, it should be noted that the CR rates and duration of the remissions are longer in the group treated initially compared with the observation groups. If one withholds treatment until a clinical indication emerges, the median time to initiation of chemotherapy is between 2 and 3 years. In one study, about one-fifth of the patients in the observation protocol remained free from an indication to begin chemotherapy at 10 years. In a large prospective British trial, which had a 19-year follow-up, the actuarial survival without needing treatment in elderly patients was 40% (out of 51 patients). If one is to choose the watchful waiting approach, careful patient selection is essential. The presence of B symptoms, peripheral blood cytopenia, kidney or liver involvement, and any life-endangering organ involvement suggest a more aggressive disease requiring immediate treatment.
A single agent oral alkylating agent like chlorambucil or cyclophosphamide may be an attractive choice in the elderly because of ease of administration and relatively low toxicity. When used alone, this therapy results in response rates of 60% to 80% with about half of all patients achieving a complete response. Combination chemotherapy with cyclophosphamide, vincristine, and prednisone (CVP) is also effective and may result in slightly higher CR of 60% to 70%. Fludarabine, a purine nucleotide analog, is also an active agent used in the treatment of indolent lymphoma. Used alone, it can induce remissions in about 60% to 80% of all treatment naive patients.
The use of interferon for treatment of indolent lymphoma remains controversial. A meta-analysis demonstrated usefulness as a maintenance therapy in patients treated with aggressive combination chemotherapy who had responded to the therapy. It was shown that the DFS was increased with addition of interferon alfa as a maintenance agent. However, many patients on interferon have symptoms of fatigue and depression that affect their quality of life.
When used as a single agent in previously untreated patients, rituximab produces response in 47% to 73% of patients with up to 37% achieving a CR. Moreover, when combined with combination chemotherapy such as CHOP, rituximab has shown an excellent overall response rate (100%) and CR (87%). When combined with CVP (cyclophosphamide, vincristine, prednisone), R-CVP yields better overall survival (81% vs. 50%), CR (57% vs. 10%), and time to progression (median 27 months vs. 7 months) than CVP alone.
Patients with advanced stage disease including involvement of extranodal sites should be informed that recent advances in treatment appear to be associated with longer remissions and longer survival, but definitive evidence of disease curability is still scarce. Formerly, CRs could be achieved with a wide range of interventions including radiation therapy, single agent and combination chemotherapy, antibodies, interferon, and various combinations. However, the CRs tended to be short-lived, with a median duration of 2 years. More active regimens and agents are changing this picture. Combination chemotherapy programs including CHOP (see earlier in the chapter) or fludarabine with rituximab produce complete responses in 90% or more of patients with median durations of complete responses exceeding 8 years. Because patients with FL can survive for many years with lymphoma, treatment-induced improvement in overall survival has been more difficult to demonstrate. However, some evidence of treatment benefit is beginning to emerge from studies that use effective drugs with rituximab during induction therapy and some sort of maintenance therapy (either rituximab or interferon). It is not clear if maintenance therapy is merely keeping disease at a subclinical level or is eradicating disease. Another benefit from aggressive treatment approaches seems to be a reduction in the rate of histologic progression.
Once a relapse occurs, the disease is likely to respond to treatment again, but the response rates and duration decrease with each relapse. Some data support the use of high-dose therapy with autologous stem cell transplant in selected (usually younger) patients. Two radionuclides have been approved by the FDA in the treatment of rituximab refractory indolent NHL. Yttrium-90-conjugated ibritumomab tiuxetan and iodine-131-conjugated tositumomab produce responses in two-thirds to three-fourths of rituximab-resistant patients. 131I tositumomab is also effective as a single agent in previously untreated patients with over 90% OR and 75% CR. However, use of these agents as a primary therapy requires further randomized studies.
The confusion in the field is evident when expert recommendations range from watch and wait to high-dose therapy with transplant. However, a reasonable approach is to treat all patients with stage I or II disease with curative intent with radiation therapy. Patients with advanced disease should be assessed for intercurrent illness and life expectancy. Patients whose life expectancy is greater than 10 years may be candidates for an initial attempt at inducing durable remission with R-CHOP. Those with a shorter life expectancy with intercurrent illness can be observed and treated palliatively with single agent oral chlorambucil with or without prednisone when needed to control symptoms.
Small lymphocytic lymphoma (SLL) is the tissue counterpart of chronic lymphocytic leukemia (CLL) and occurs in patients with a median age of 65 years. CLL is the most common form of leukemia in the United States (Figure 104-1
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


