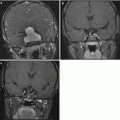
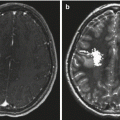
Fig. 8.1
Ganglioglioma. (a) T1-weighted axial MRI with gadolinium of a ganglioglioma in the left temporal lobe. Nodular enhancement is seen. (b) Hematoxylin and eosin staining reveals large, dysplastic neurons and a neoplastic glial component. Necrosis is not seen
The 4th edition of the WHO classification does not include grade II as a designation for gangliogliomas (Louis et al. 2007). In this edition, gangliogliomas are designated WHO grade I and anaplastic gangliogliomas are designated WHO grade III. Grade III neoplasms are characterized by additional features such as atypia (increased cellularity, conspicuous pleomorphism), microvascular proliferation, or an elevated MIB-1 labeling index. Necrosis is absent unless the glial component undergoes malignant transformation. Tumors with evidence of malignant transformation are considered anaplastic gangliogliomas, WHO grade III.
8.2.2.3 Immunohistochemistry and Electron Microscopy
Immunohistochemical staining techniques are crucial for identifying the neuronal and astrocytic features within these tumors. Positive staining for synaptophysin, neuropeptides, and biogenic amines are associated with a neuronal phenotype. Similarly, positive staining for glial fibrillary acid protein (GFAP) identifies the astrocytic component. Electron microscopy is also helpful to identify additional neuronal features such as dense core granules and synaptic junctions (Hirose et al. 1997; Gelabert-Gonzalez et al. 2010).
8.2.2.4 Cytogenetics
Molecular cytogenetic data regarding ganglioglioma are scarce, but a few abnormal karyotypes have been observed. Specific cytogenetic abnormalities include a ring chromosome 1, trisomy of chromosomes 5–7, and deletion of chromosome 6 (Neumann et al. 1993; Xu et al. 2014). Analysis for microsatellite marker instability in tumor DNA from six gangliogliomas found no abnormalities (Zhu et al. 1996). One series of ganglioglioma patients reported a comparatively higher frequency of splice-site-associated single-nucleotide polymorphism in the tuberous sclerosis 2 gene (TSC2) (Platten et al. 1997). This may suggest an underlying genetic susceptibility for sporadic ganglioglioma, although the underlying biologic mechanism is unknown. More recently described variants, papillary glioneuronal tumor and rosette-forming glioneuronal tumor (WHO grade I), have been mostly described in adults, although a recent systematic review also identified a number of children with these tumors (Schlamann et al. 2014). Chromosomal and structural alterations involving only chromosome 7 with breakpoints at 7p22 have been reported in the papillary glioneuronal tumor variant (Faria et al. 2008).
8.2.3 Clinical Features
Seizure is the most common presenting symptom of gangliogliomas. The seizure history is often longstanding, with a mean duration prior to diagnosis ranging from 6 to 25 years (Prayson et al. 1995; Luyken et al. 2003; Southwell et al. 2012). In one series of patients, temporal lobe gangliogliomas represent 40 % of all tumors causing chronic temporal lobe epilepsy (Blumcke et al. 1999). Patients with brain stem lesions commonly present with involvement of the motor tracts: weakness, spasticity, and gait disturbance (Gopalakrishnan et al. 2013). Gangliogliomas of the spinal cord may involve the entire spinal cord and typically produce scoliosis, gait disturbance, and progressive weakness (Hamburger et al. 1997; Jallo et al. 2004). These symptoms can be very longstanding before a diagnosis is reached. Patients with midline tumors may develop symptoms and signs of hydrocephalus, such as headache, papilledema, alterations in the level of consciousness, and nausea/vomiting (Haddad et al. 1992; Deling et al. 2013; Zhang et al. 2013a).
8.2.4 Natural History
Gangliogliomas are indolent, slow-growing tumors. Without resection, patients often have prolonged courses of disease, depending on the location of the primary mass (Selch et al. 1998). Anaplastic glial changes in ganglioglioma, as well as high MIB-1 labeling indices, may be markers for more aggressive tumor behavior (Hirose et al. 1997; Scoccianti et al. 2012). Malignant transformation is relatively rare, occurring in less than 3 % of gangliogliomas (Hakim et al. 1997; DeMarchi et al. 2011).
8.2.5 Diagnosis and Neuroimaging
Neuroimaging is the initial step in diagnosis, as no specific laboratory tests are available. A CT scan, if performed as a screening test, reveals an iso- to hypodense solid or cystic mass (Adachi and Yagishita 2008). Cysts may be associated with a mural nodule, although both cyst and nodule are well circumscribed. Calcifications may be present and contrast enhancement is usually seen, but occasionally can be minimal or absent (Lagares et al. 2001). MRI is the best imaging modality and is required to adequately delineate the mass. Tumors are usually hypointense on T1-weighted images and hyperintense on T2-weighted images (Zhang et al. 2008). Mass effect and edema are typically minimal. Contrast enhancement varies in intensity and may be nodular, rim-like, or entirely solid (Figs. 8.1a and 8.2). Syringobulbia or syringomyelia can be seen with spinal cord gangliogliomas (Park et al. 1993; Hamburger et al. 1997; Jallo et al. 2004). MR spectroscopy is usually of limited value due to the indolent nature of these tumors.
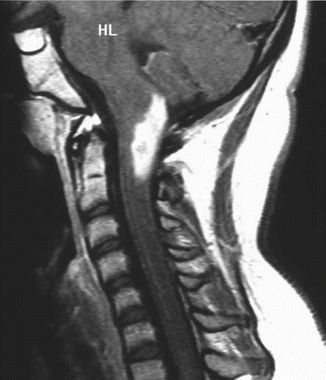
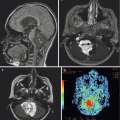
Fig. 8.2
An unusual case of a ganglioglioma of the upper cervical spinal cord. The patient is a 14-year-old girl who presented with paresthesias over the left side of the neck. The sagittal T1-weighted post-contrast MRI shows a well-demarcated mass arising in the dorsal portion of the spinal cord
8.2.6 Treatment
Complete surgical resection is the treatment of choice and when achievable is usually curative (Sutton et al. 1987; Compton et al. 2012). The neoplasm itself contains no functioning nervous tissue, but potential eloquence of surrounding parenchyma must be considered in surgical planning (Southwell et al. 2012). A postoperative MRI is useful to assess the extent of resection. Subsequent surveillance imaging should be done to evaluate for recurrence, which can occur in a small percentage of patients. Radiation therapy should be considered for tumor recurrence when further resection is not feasible. Tumors with malignant features (anaplastic features, high MIB-1 labeling index) may require radiation therapy as an adjuvant therapy, regardless of the extent of resection (Hakim et al. 1997; DeMarchi et al. 2011). Radiation therapy for benign lesions may delay time to progression in patients with unresectable disease, but the impact of radiotherapy on progression-free survival for incompletely resected benign tumors remains uncertain (Lang et al. 1993; Compton et al. 2012). Because the role of radiation therapy for subtotally resected, low-grade tumors is unclear, the risks and benefits of radiotherapy should be carefully weighed.
8.2.7 Outcome
The prognosis following GTR is excellent (Khajavi et al. 1995; Compton et al. 2012; Englot et al. 2012a; Southwell et al. 2012). In one surgical series of 88 patients with a median follow-up of nearly 12 years, 15-year overall survival was 94 % and 10-year progression-free survival was 37 %, with progression being dramatically affected by the extent of the initial resection (Compton et al. 2012). Tumor location is a significant predictor of outcome, most likely because it predicts resectability. Patients who undergo a subtotal excision (STR), most commonly seen in patients with midline tumors, are at higher risk of tumor progression or recurrence (Haddad et al. 1992; Compton et al. 2012). The importance of anaplasia as a prognostic feature is unclear, with different series demonstrating conflicting results (Kalyan-Raman and Olivero 1987; Lang et al. 1993; DeMarchi et al. 2011; Selvanathan et al. 2011). A retrospective analysis in one series of 34 patients, however, did demonstrate a correlation between improved survival and degree of resection as well as tumor grade (Selch et al. 1998). In a large series reported by Luyken et al., the rate of 7.5-year, progression-free survival was 97 % (Luyken et al. 2003). Risk factors for recurrence or malignant progression were residual tumor, frontal tumor location, and a higher-grade lesion. The survival outcomes are also acceptable for gangliogliomas involving the posterior fossa (Baussard et al. 2007) or spinal cord (Jallo et al. 2004).
For patients with tumor-associated epilepsy, seizure control improves dramatically after tumor resection (Englot et al. 2012a; Wallace et al. 2013). GTR appears to be the most important treatment-related factor (Benifla et al. 2006; Giulioni et al. 2006; Park et al. 2008; Southwell et al. 2012). In a recent series of 66 patients with ganglioglioma, 49 of which presented with epilepsy, and 85 % of patients with a seizure history were free of seizures after surgery (Southwell et al. 2012). Ninety-six percent of individuals in whom GTR was achieved were seizure-free, but only 54 % of the group had STR. A recent systematic review and meta-analysis found that postoperative seizure freedom in glioneuronal tumor resection was predicted by GTR, the absence of generalized seizures, and a shorter history of epilepsy (Englot et al. 2012a). Because seizure outcomes are improved in those patients with a shorter duration of epilepsy, early surgical intervention should be considered, particularly in patients with medically refractory epilepsy.
8.3 Dysembryoplastic Neuroepithelial Tumor
Dysembryoplastic neuroepithelial tumor (DNET) is a benign glioneuronal neoplasm that most commonly occurs in the supratentorial compartment. It was first described by Daumas-Duport and Scheithauer in 1988 (Daumas-Duport et al. 1988). The initial report described 39 children with a morphologically distinct brain tumor and intractable partial seizures.
8.3.1 Epidemiology
DNET most commonly affects children and young adults in the second and third decade of life, with a peak in late childhood to early adolescence (Daumas-Duport and Varlet 2003). The incidence of DNET is not accurately known, but available reports suggest it affects 0.6–3 % of individuals with a primary brain tumor (Morris et al. 1993; Wolf et al. 1995; Rickert and Paulus 2001; Rashidi et al. 2003). In a retrospective review of all neuroepithelial tumors at a single institution, DNETs were found in 0.6 % of patients including all ages, in 1.2 % of patients under age 20 years, and in 0.2 % of patients over 20 years of age (Daumas-Duport et al. 1988). Males are more frequently affected than females (Daumas-Duport et al. 1988; Rickert and Paulus 2001).
8.3.2 Pathology
8.3.2.1 Gross Appearance
DNET arises from and expands the cortex, although the underlying white matter may also be involved, and it has traditionally been viewed as a benign “quasihamartomatous” tumor (Daumas-Duport 1993; Ray et al. 2009). Distended cortical ribbons consisting of gelatinous glioneuronal elements and smaller, firmer glial nodules are seen during surgery.
8.3.2.2 Histopathology
The characteristic pathologic feature is the glioneuronal element which consists of columns of axon bundles lined with small S100-positive and GFAP-negative oligodendroglia-like cells (Fig. 8.3b). Oligodendroglia-like cells have minimal cytoplasm and are rich in mucopolysaccharides. Mature neuronal cells are found interspersed within the tumor and adjacent cortical dysplasia can be found. Associated cortical dysplasias are commonly observed with DNET (Fay-McClymont et al. 2012).
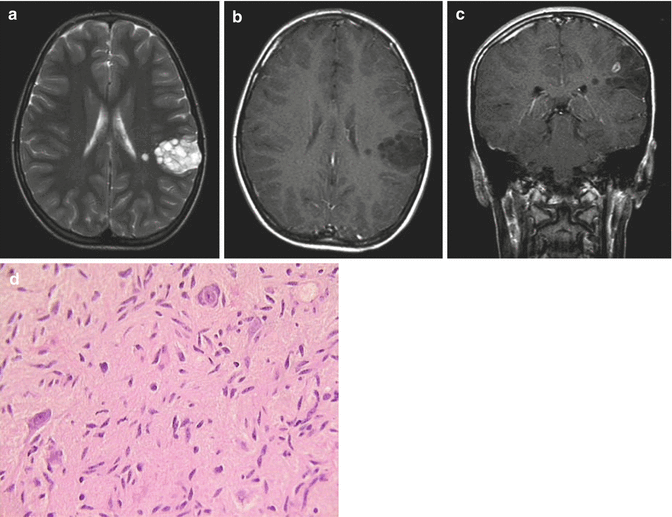
Fig. 8.3
Dysembryoplastic neuroepithelial tumor (DNET). (a) T2-weighted axial MRI of a left parietal DNET demonstrates a typical “bubbly” appearance. The majority of the mass does not enhance following contrast (b), although a small focus of enhancement was noted along one margin of the tumor (c). No peritumoral edema is seen. (d) H & E staining in a complex form of DNET shows nuclear atypia of glioneuronal elements. Astrocytic, oligodendrocytic, and neuronal components are present to varying degrees
Smaller glial nodules are found along the tumor borders of the complex variant of DNETs. In contrast to gangliogliomas, atypical neurons resembling ganglion cells and perivascular lymphocytes are not found with DNETs (Daumas-Duport 1993; Hirose et al. 1994; Raymond et al. 1994; Fay-McClymont et al. 2012). DNETs are classified as WHO grade I, and although rare, malignant transformation has been observed (Ray et al. 2009; Mano et al. 2013).
8.3.2.3 Immunohistochemistry and Molecular Genetics
Neuronal elements stain positive for synaptophysin and neuronal nuclear antigen (NeuN) (Wolf et al. 1997; Brandes et al. 2000). Glial nodules stain positive for GFAP. The proliferation potential is very low and MIB-1 labeling indices vary from 0 to 8 % (Prayson and Estes 1992; Daumas-Duport 1993; Taratuto et al. 1995). Molecular genetic abnormalities have not been well studied, but one study identified certain tumors with IDH1 mutation, 1p/19q loss, isolated loss 9q, and/or PTEN loss, which were not associated with tumor type or location or higher cell proliferation (Fay-McClymont et al. 2012). DNETs have been reported in patients with neurofibromatosis type I, although the overall frequency is unknown, and the lack of NF1 gene loss in some of these tumors puts into question whether they are truly associated with defects in NF1 expression (Lellouch-Tubiana et al. 1995; Fedi et al. 2004).
8.3.3 Clinical Features
DNETs are associated with chronic, intractable partial seizures and are present in 25 % of all lesions resected for medically refractory epilepsy (Wolf et al. 1995; Pasquier et al. 1996; Chang et al. 2010). Most DNETs are located in the supratentorial region, especially the temporal lobe; however, other locations corresponding to the topography of the secondary germinal layers have been described, including the basal ganglia, thalamus, cerebellum, and pons (Leung et al. 1994; Kuchelmeister et al. 1995; Cervera-Pierot et al. 1997). Multifocal locations have been described, including both supratentorial and infratentorial (including both the cerebellum and brain stem) lesions in the same patient (Leung et al. 1994; Sharma et al. 2009).
8.3.4 Natural History
Untreated lesions often do not grow, but without resection, medically intractable seizures are likely to persist (Chang et al. 2010; Thom et al. 2011). Tumor progression is rare with partially resected DNETs, but does occur, and may suggest malignant transformation (Daumas-Duport 1993; Raymond et al. 1994; Taratuto et al. 1995; Mano et al. 2013; Kim et al. 2014). Subtotally resected lesions can remain quiescent for extended periods of time.
8.3.5 Diagnosis and Neuroimaging
Appearance on unenhanced CT ranges from iso- to hypodense, often with calcifications and occasionally with true cyst formation. One-third of tumors show contrast enhancement and the overlying calvarium may be remodeled, consistent with the chronic nature of the tumor (Daumas-Duport 1993; Raymond et al. 1995; Stanescu Cosson et al. 2001). DNETs are cortically based and may appear as macrogyri. Usually, the lesion involves the thickness of the normal cortex, although it can extend into the white matter. With MR imaging, the tumor is hypointense on T1-weighted and hyperintense on T2-weighted images (Fig. 8.3). No peritumoral edema or mass effect is usually seen. Enhancement is seen in one-third of tumors (Fig. 8.3c) (Daumas-Duport 1993; Raymond et al. 1995; Campos et al. 2009; Mano et al. 2013).
A definitive diagnosis of DNET is difficult to obtain with neuroimaging alone. However, the combination of partial seizures before age of 20 years, lack of progressive neurologic deficit, cortical involvement on MRI, absence of mass effect, or edema on CT or MRI is highly suggestive of DNET (Daumas-Duport 1993; Lang et al. 1993; Fernandez et al. 2003).
8.3.6 Treatment
Surgical GTR is typically curative. Recurrence has been reported rarely; therefore, radiation or chemotherapy is usually not indicated, except in cases of rare malignant lesions (Raymond et al. 1995; Maher et al. 2008; Mano et al. 2013). It is important to differentiate DNET from oligodendroglioma to avoid unnecessarily aggressive therapy.
8.3.7 Outcome
Neither clinical nor radiographic tumor progression is seen in the majority of patients, even with subtotal resection (Daumas-Duport et al. 1988; Daumas-Duport 1993; Raymond et al. 1994; Taratuto et al. 1995; Chang et al. 2010). Resection results in an approximately 83 % rate of seizure control across the literature, as observed in a recent systematic review (Englot et al. 2012a). In one report of 50 patients with DNET-related epilepsy, 87 % achieved seizure freedom after surgery (Chang et al. 2010). Seizure freedom was predicted by GTR, achieved in approximately 80 % of surgeries, and this outcome remained resilient at a median follow-up of greater than 5 years. Currently, no agreement exists over whether removal of the tumor alone (lesionectomy) or extended resection to include neighboring dysplastic cortex results in the best seizure control (Nolan et al. 2004; Chan et al. 2006; Giulioni et al. 2006; Minkin et al. 2008; Englot et al. 2012b).
8.4 Central Neurocytoma
8.4.1 Epidemiology
Central neurocytomas are rare CNS neoplasms and compromise only 0.25–0.5 % of brain tumors and are tumors of adolescents and young adults (Hassoun et al. 1993; Yang et al. 2015). In one series of 207 cases, the mean age of presentation was 29 years, with a range of 8 days to 67 years (Hassoun et al. 1993). Approximately 70 % of patients present between the ages of 20 and 40 years, and the incidence is similar in males and females (Vasiljevic et al. 2013).
8.4.2 Pathology
8.4.2.1 Gross Appearance
Central neurocytomas are lobulated, well-circumscribed masses that are gray in color, similar to the normal cortex (Bonney et al. 2015). They typically occur in close proximity to the foramen of Monro and may be attached to the septum pellucidum. Necrosis and cyst formation are frequently seen, and some neurocytomas are very vascular. Intratumoral hemorrhage is unusual.
8.4.2.2 Histopathology
The histopathologic appearance of a central neurocytoma can be similar to that of an oligodendroglioma (von Deimling et al. 1990; Schild et al. 1997; Bonney et al. 2015). Both neoplasms have small uniform cells with rounded nuclei and scant cytoplasm resembling perinuclear halos (the so-called “fried egg” appearance). It is quite likely that many intraventricular tumors previously diagnosed as oligodendrogliomas may actually have been central neurocytomas (von Deimling et al. 1990; Schild et al. 1997). The cytoplasm is ill defined and the nuclei are round to slightly lobulated (Fig. 8.4b). The tumor cells are dense in some areas and alternate with anuclear, less dense tumor parts. In particular the anuclear areas may have a fine fibrillary matrix. A delicate pattern of blood vessels forms a branching network in pattern similar to oligodendrogliomas. Focal calcification can be seen. Mitotic figures are absent or infrequent and endothelial proliferation and necrosis are uncommon. A variant, extraventricular neurocytoma (WHO grade II) usually occurs in adults and can sometimes be difficult to distinguish from oligodendroglioma (Mut et al. 2005; Sweiss et al. 2015).
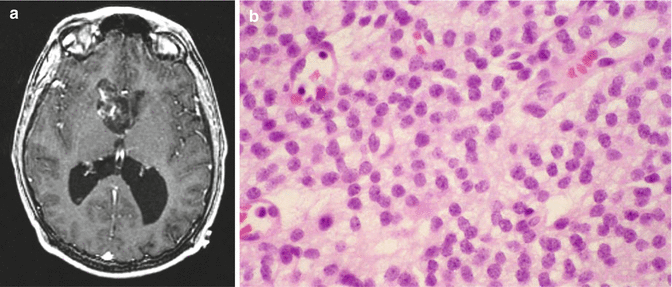
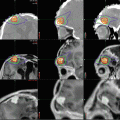
Fig. 8.4
Central neurocytoma. (a) T1-weighted axial MRI with gadolinium of a typical central neurocytoma arising in the frontal horn of the right lateral ventricle. A heterogeneous pattern of enhancement is seen. (b) H & E staining demonstrates cells with uniform, round to oval nuclei with speckled chromatin and occasional nucleolus. Anaplastic features are not seen
8.4.2.3 Immunohistochemistry and Electron Microscopy
Immunostaining for neuron-specific enolase (NSE) and synaptophysin confirms the neuronal origin of these tumors (von Deimling et al. 1991; Bonney et al. 2015). Positive staining with GFAP may represent neoplastic or reactive astrocytes. It has been suggested that central neurocytomas originate from bipotential (neuronal and astrocytic) progenitor cells in the periventricular region which persist into adulthood (von Deimling et al. 1991). An ultrastructural feature that sometimes distinguishes central neurocytomas from oligodendroglioma is the high degree of neuronal maturation. Electron microscopy demonstrates clear and dense-core vesicles, microtubules, and synapse formation.
8.4.2.4 Cytogenetics and Molecular Genetics
Comparative genomic hybridization (CGH) analysis was used to identify losses and gains in DNA sequences in ten histologically confirmed central neurocytomas (Yin et al. 2000). Genomic alterations were found in six tumors. Gain in genetic material was found for chromosomes 2p and 10q in four tumors, chromosome 18q in three tumors, and in chromosome 13q in two tumors. Gains in chromosome 7 were reported in three out seven central neurocytomas using fluorescence in situ hybridization (FISH) (Taruscio et al. 1997). Loss of 1p/19q has been described in a subset of extraventricular but not intraventricular neurocytomas and may be associated with aggressive histology in these tumors (Rodriguez et al. 2009).
It has been proposed that central neurocytoma originates from an adult neuronal progenitor cell. A significant overlap in the antigen profile and gene expression was observed in tumor specimens and native neuronal progenitor cells. GDF8, PDGF-D, neuregulin 2 (NRG2), IGF2, and JAG1 were overexpressed in tumors, suggesting that central neurocytoma is characterized by the concurrent overactivation of these pathways, which may drive neurocytoma expansion, while restricting tumor progenitor phenotype (Sim et al. 2006). One recent report identified genes highly expressed in five neurocytomas compared to the normal brain, including several in the Wnt/β-catenin and sonic hedgehog (SHH) signaling pathways, as well as genes mainly linked to calcium function or maintenance of neural progenitors (Vasiljevic et al. 2013).
8.4.3 Clinical Features
Patients most often present with symptoms attributable to raised intracranial pressure secondary to obstructive hydrocephalus (Yang et al. 2015). As expected, these consist of headaches and visual changes; the duration of clinical symptoms and signs is typically less than 6 months. In one study, 93 % of patients complained of headaches, 37 % had visual changes, and 30 % experienced nausea and vomiting at presentation, while individuals less commonly complained of paresthesias (19 %), lethargy (11 %), balance problems (11 %), and tinnitus (7 %) (Schild et al. 1997). On physical exam, common presenting signs include papilledema and ataxia (Schild et al. 1997; Yang et al. 2015).
8.4.4 Natural History
While most central neurocytomas are benign, they can recur and even disseminate along the CSF pathways (Eng et al. 1997; Kim et al. 2013). Anaplasia has been demonstrated in central neurocytomas, marked by increased proliferative potential, and it is associated with worse long-term survival and local tumor control (Choudhri et al. 2015). An increase in GFAP positivity and vascular proliferation may suggest a more malignant course (Elek et al. 1999).
Most reports indicate that central neurocytomas are relatively slow growing, with the exception of atypical variants (Soylemezoglu et al. 1997; Sharma et al. 1998; Choudhri et al. 2015). Markers of proliferation have been studied in order to clarify the biological behavior of neurocytomas. In one study of 36 central neurocytomas, it was found that MIB-1 index under 2 % had a 22 % relapse rate, compared to a relapse rate of 63 % when the MIB-1 index was over 2 % for the observation period of 150 months (Soylemezoglu et al. 1997). With longer follow-up, tumors with an increasing MIB-1 index may relapse. This is illustrated in a case report of a patient with a recurrent central neurocytoma that had a fourfold increase in MIB-1 index after a 9-year disease-free interval (Christov et al. 1999). Necrosis and increased mitotic figures have also been reported in tumors with high-growth potential (Choudhri et al. 2015). Atypical (or anaplastic) neurocytomas (either in an intra- or extraventricular location) are now typically defined as having a high proliferative index (MIB-1 index >2 %) and may also have vascular proliferation, increased mitotic figures, and necrosis (Choudhri et al. 2015).
8.4.5 Diagnosis and Neuroimaging
CT scans demonstrate an iso- or slightly hyperdense mass within the body of the lateral ventricles near the foramen of Monro (Donoho and Zada 2015). Areas of hypodensity represent cystic degeneration. About one-half of central neurocytomas demonstrate calcification on CT imaging (Hassoun et al. 1993). These tumors are thought to arise from septal nuclei and have broad-based attachments to the superior and lateral walls of the ventricle. Obstruction of the interventricular foramen of Monro by tumor mass usually results in hydrocephalus. Contrast enhancement is mild to moderate for most central neurocytomas.
MRI reveals an isointense mass on T1-weighted images, with a soap-bubble multicystic appearance on T2-weighted images (Donoho and Zada 2015). Most central neurocytomas are isointense on T2-weighted images. Moderate heterogeneous gadolinium enhancement is seen (Fig. 8.4a) (Wichmann et al. 1991; Donoho and Zada 2015). Catheter angiography is rarely performed for central neurocytomas, but if obtained shows a homogenous vascular blush. On occasion, tumors can be relatively avascular (Taratuto et al. 1995; Ashkan et al. 2000). Arterial supply is from the posterior and anterior choroidal, pericallosal, and lenticulostriate vessels.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


