Chapter Outline
Development of the Sympathetic Nervous System
Genetic Disorders Associated with Neuroblastoma
Hereditary Predisposition to Neuroblastoma
Genetic Aberrations in Sporadic Neuroblastoma
Chromosomal Abnormalities in Neuroblastoma
Neurotrophin Receptors in Neuroblastoma
Treatment of Intermediate-Risk Disease
Treatment of High-Risk Disease
Treatment of Minimal Residual Disease
Assessment of Response to Treatment
Neuroblastoma is an embryonal tumor of the sympathetic nervous system arising from the neural crest. It is a common extracranial solid tumor of childhood and is a disease distinguished by its clinical and biologic heterogeneity. The prognosis for persons with neuroblastoma is variable and largely dependent on tumor biology. Patients with localized disease and favorable tumor biology may be successfully treated with surgery alone or with minimal therapy with an estimated 5-year survival rate of 90%. However approximately half of all patients present with metastatic disease, adverse tumor-specific biologic features, or both. For these children with “high-risk” disease features, cure rates remain poor, with estimated 5-year survival rates of 50%. The development of resistance to chemotherapy and radiation is the cause of most treatment failures, and neuroblastoma accounts for 10% of childhood cancer mortality. This chapter summarizes our current understanding of neuroblastoma biology and pathogenesis and how this rapidly evolving insight has affected current and future treatment strategies.
Epidemiology
Neuroblastoma accounts for 7% of all childhood cancer diagnoses, with more than 650 new cases diagnosed in the United States each year. The incidence of neuroblastoma is 10.5 per million children younger than 15 years of age per year. It is the most commonly diagnosed cancer of infancy. The median age of diagnosis is 19 months, with a peak incidence in children younger than 4 years. There is no racial preponderance, although African Americans are more likely to have high-risk disease and a fatal outcome. Large-scale genetic studies to investigate how this likelihood may influence the disease phenotype revealed that a single nucleotide polymorphism (SNP) within the sperm-associated antigen 16 (SPAG16) gene had a higher risk allele frequency in the African reference population and contributed to the ethnic disparities in high-risk disease and survival.
The cause of neuroblastoma is largely unknown. The embryonal origin of the tumor, as well as the young age of onset, suggest that prenatal and perinatal exposures may be important. Studies have investigated a variety of prenatal exposures including tobacco, alcohol, pesticides, and maternal medication or drug use, as well as birth characteristics, including small size for gestational age and maternal history of fetal loss. The findings from these studies have been inconsistent, and none of these associations has been confirmed in large studies.
Because the majority of neuroblastomas produce catecholamine metabolites that can be detected in the urine, infant screening programs have been conducted to see if it is possible to decrease the mortality from high-risk neuroblastoma by diagnosing cases earlier. In Japan, starting in 1985, a nationwide mass screening program for neuroblastoma was conducted in 6-month-old infants. Results showed that the incidence rate of neuroblastoma increased two- to threefold as a result of the screening, implying that without screening, many of these tumors would have regressed spontaneously and would never have been diagnosed clinically. In addition, the majority of patients had low-stage disease and biologically favorable features, and screening did not decrease overall neuroblastoma mortality. Through the Quebec Neuroblastoma Screening Program, infants were screened at 3 weeks and 6 months; the findings confirmed the Japanese results and showed a significant complication rate in patients undergoing treatment for tumors found by screening. In the German Neuroblastoma Screening Study, investigators looked at postponing screening until 10 to 19 months of age. They found less “overdiagnosis” of neuroblastoma and a greater frequency of patients with unfavorable clinical and biologic features. However, no decrease in the mortality of the patients with unfavorable features was documented. Because of these results, mass screening efforts for neuroblastoma in infants have largely been abandoned.
Pathogenesis
Embryology
Neuroblastoma arises during development from neural crest cells committed to the sympathoadrenal lineage. The two predominant cell types comprising neuroblastoma are neuroblasts and Schwann cells. Neuroblasts derive from pluripotent cells that arise in the neural crest and eventually form various components of the sympathetic nervous system: the sympathetic ganglia, the chromaffin cells of the adrenal medulla, and the paraganglia, which are typical sites where neuroblastic tumors can arise. What causes persistence of these embryonal cells that develop into peripheral neuroblastic tumors is unclear. Genes that dictate neural development could be altered, causing defects in the normal differentiation and programmed cell death pathways, leading to uncontrolled proliferation and aberrant cell survival.
Microscopic neuroblastic nodules have been reported in fetal adrenal glands during development, peaking at around 17 to 20 weeks’ gestation and regressing by birth or within the first few months of postnatal life. Beckwith and Perrin (1963) termed these nodules, which were histologically identical to neuroblastoma, “neuroblastoma in situ,” which they noted during postmortem examinations of infants younger than 3 months of age who died of other causes. These lesions were detected with a frequency fiftyfold higher than the expected incidence of primary adrenal neuroblastoma and were initially thought to be neuroblastomas that regressed spontaneously. However, with new insights into the development of the adrenal gland, it is clear that these are remnants from normal fetal development. Indeed, the genetic profiles of normal developing neuroblasts and malignant neuroblastomas are similar in many respects, as shown by messenger ribonucleic acid (mRNA) expression profiling of sympathetic neuroblasts from human fetal adrenal glands.
Development of the Sympathetic Nervous System
The sympathetic nervous system is derived from the neural crest, a multipotent embryonic structure that appears early in development and is made up of a transient population of cells that migrate from the neural tube to the region of the dorsal aorta during development. These cells develop into sympathetic, parasympathetic, enteric, or sensory neurons governed by transcriptional regulators and growth factors as they exit from the neural tube, during migration or at sites where these cells differentiate. Development of the sympathetic phenotype is summarized in Figure 54-1 . Premigratory neural crest cells are specified by the expression of several markers, including FoxD3 and SOX10. Foxd3 is required for sublineage fate specification, migration, and survival. Zebrafish homozygous for foxd3 inactivating mutations have nearly a complete loss of sympathetic neurons and their precursor populations. Sox10 is responsible for the multipotency of neural crest–derived cells and is required for sympathetic nervous system and melanocyte development. At the dorsal aorta, prespecified SOX10 -positive sympathetic progenitors that are under the influence of extrinsic factors, bone morphogenetic proteins, start the process of differentiation into noradrenergic neurons. The basic helix-loop-helix (bHLH) transcription factor Ascl1 and the homeodomain transcription factor Phox2b are the first transcription factors that are expressed upon initiation of differentiation of sympathoadrenal precursors. Phox2a , a homolog of PHOX2B , is expressed downstream of both Phoxb and Ascl1, but its role in the initial stages of the sympathetic lineage remains undefined. Further neuronal differentiation occurs under the influence of the transcription factors Hand2 , Gata2/3, and Tfap2a , which interact in a complex regulatory network to ultimately induce the expression of tyrosine hydroxylase (TH) and dopamine beta hydroxylase. These two enzymes are required for catecholamine production and serve as markers of terminal sympathetic neuronal differentiation. Hand2 appears to have more of a role in noradrenergic cell proliferation than differentiation. A massive reduction in sympathetic ganglion size occurs after elimination of Hand2 in the neural crest of model organisms because of a nearly complete block in proliferation of immature sympathetic neurons. Thus different members of the gene regulatory network appear to have selective functions, some in proliferation and others in differentiation.
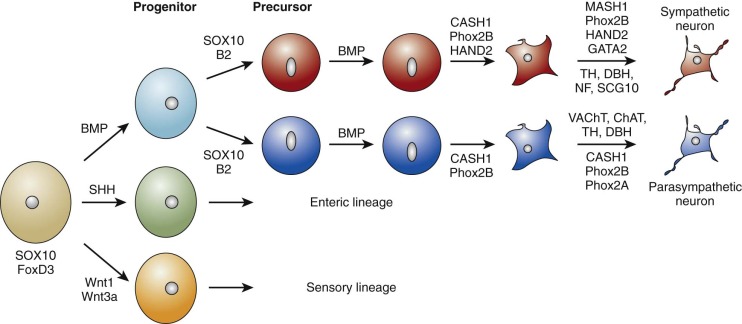
Neural crest cells from the cranial region migrate from the neural folds into the pharyngeal arches and heart, where they play essential roles in septation of the single outflow of the heart (the truncus arteriosus) and in the formation of the conotruncal portion of the ventricular septum. It follows that coexistence of neuroblastoma with anomalies of other neural crest–derived structures is biologically plausible because of the shared roles of regulatory and developmental genes. For example, ablation of the cardiac neural crest results in congenital cardiovascular malformations in the chick embryo. Indeed, a significantly increased incidence of congenital cardiac malformations, especially those that involve neural crest–derived tissues, was noted in patients with neuroblastoma compared with other malignancies, such as leukemia. These cardiac anomalies were seen primarily in patients with neuroblastoma who were younger than 1 year and had lower stage tumors. Menegaux et al. (2005) confirmed the association of neuroblastoma with conotruncal cardiac anomalies, as well as with various urogenital anomalies; other studies have shown varying results. Although the malignant transformation of neuroblasts and the disruption of normal cardiogenesis are likely to be multistep processes, it is possible that shared initiating genetic lesions occur very early in development, causing derangement of common developmental pathways.
Genetic Disorders Associated with Neuroblastoma
Neuroblastoma may occur rarely in association with chromosomal anomalies. In addition to the increased incidence of neuroblastoma in girls with Turner syndrome, neuroblastoma has also been noted to occur in patients with Noonan syndrome. The gene encoding the protein phosphatase PTPN11 is mutated in 50% of patients with Noonan syndrome and is associated with an increased risk of leukemia and neuroblastoma. Other neuro-cardio-facial syndromes such as Costello syndrome have been associated with neuroblastic tumors. Mutations in the neurofibromatosis type 1 (NF1) gene have been detected in neuroblastoma cell lines, and the literature includes one case report of a patient with a germline NF1 mutation and neuroblastoma who had a homozygous deletion of the NF1 gene in the tumor. However, germline inactivation of Nf1 does not predispose to neuroblastoma in the mouse.
Persons affected with overgrowth disorders such as Beckwith-Weidemann syndrome are at risk for embryonal tumors, including neuroblastoma. The increased risk is mainly during the first 4 years of life, and current recommendations include quarterly evaluation with abdominal ultrasound until age 8 years. The molecular aberrations that predispose to neuroblastoma in particular are unknown, although maternal differentially methylated region 2 (DMR2) loss of methylation that occurs in 50% to 60% of cases is thought to confer a 1% to 5% risk of developing non–Wilm tumors, including neuroblastoma, hepatoblastoma, rhabdomyosarcoma, adrenocortical tumors, gonadoblastoma, and thyroid carcinoma. Mutations of the CDKN1C tumor suppressor gene (encoding the cyclin-dependent kinase inhibitor p57 KIP2 ), which occur in 5% to 10% of sporadic and 30% to 50% of familial cases, is associated with a 4% estimated risk of non–Wilm tumor formation. The X-linked Simpson-Golabi-Behmel syndrome type 1 (SGBS1), associated with mutations in glypican 3 and 4 (GPC3 and GPC4) genes, is characterized by prenatal and postnatal macrosomia, distinctive craniofacies with palatal anomalies, congenital heart defects, and neuroblastoma (reviewed in reference ). Surveillance is recommended for embryonal tumors starting in the newborn period.
Neuroblastoma Stem Cells
The natural course of high-risk neuroblastoma, which results in relapse and a refractory state after an initial good response, suggests the likely existence of cancer stem cells (CSCs) in these tumors. CSCs are a subpopulation of cells within a tumor with the ability to self-renew, generate differentiated progeny, and reproduce a heterogeneous cancer population. These cells have enhanced tumorigenicity, resistance to chemotherapy, and the ability to establish metastatic foci. Hansford et al. (2007) demonstrated that dissociated cells from neuroblastoma tumors and bone marrow metastases grow as spheres and are capable of self-renewal. Spheres generated from the tumors underwent differentiation under neurogenic conditions to form neurons. Neuroblastoma stem cells from high-risk tumors expressed markers of neural crest stem cells and clinical markers of neuroblastoma, exhibited a higher self-renewal rate than those from low-risk tumors, and formed metastatic tumors that could be serially passaged in xenograft models. Compounds that selectively targeted these tumor-initiating cells (TICs)—DEC14 and rapamycin—were identified; they induced regression of TIC tumor models in vivo and markedly decreased self-renewal or tumor initiation capacity. Moreover, compounds that targeted PI3K/Akt, PKC, aurora kinase, ErbB2, Trk, and polo-like kinase–1 (PLK1) emerged from a small molecule inhibitor library screen designed to identify signaling pathways important for the survival and self-renewal of neuroblastoma TICs. Genetic and chemical inhibition of PLK1 caused regression of TIC tumor models both singly and in combination with irinotecan, suggesting that this kinase may regulate TIC growth and survival.
Cluster of differentiation (CD)–133, in addition to being a marker of hematopoietic stem cells, is also a marker of TICs in several human cancers. CD133-positive subpopulations of cells with tumor-initiating and metastatic properties have been identified in neuroblastoma tumors. Similarly, the stemness-related polycomb repressive complex–1 (PRC1) protein BMI1 is thought to have a role in tumor sphere formation. BMI1 is thought to inhibit neuroblastoma differentiation by repressing expression of the tumor suppressor in lung cancer–1 (TSLC1) and kinesin family member 1B (KIF1B) in these cells.
In a 2013 study, surface expression of the granulocyte colony-stimulating factor receptor CD114 was found to distinguish a subpopulation of neuroblastoma cells in cell lines and primary tumors, the proportion of which was increased in relapsed tumors. These cells were found to have increased responsiveness to signal transducer and activator of transcription–3 (STAT3), a critical transcription factor controlling neural crest specification. This subpopulation of cells was highly tumorigenic, could self-renew, and could give rise to differentiated progeny with characteristics of premigratory neural crest cells. This subpopulation possessed an undifferentiated phenotype similar to that found in early multipotent neural crest precursors. Further studies are required to understand the role of CSCs in neuroblastoma.
Hereditary Predisposition to Neuroblastoma
A family history has been identified in 1% of cases of neuroblastoma and is suggestive of autosomal-dominant inheritance. In these children, neuroblastoma develops at an earlier age and often manifests as multiple primary tumors.
PHOX2B Mutations
The first predisposition mutation identified in neuroblastoma was in PHOX2B . Phox2b is a key regulator of autonomic neuron development, because its complete absence leads to embryonic lethality in mice as a result of the failure of sympathetic nervous system formation. This gene is transiently expressed in murine primary sympathetic neuron progenitors from E10.5 to E13 and regulates their differentiation. Phox2b also has growth inhibitory effects, because its overexpression promotes cell cycle exit and inhibits the proliferation of cultured sympathetic neurons.
Heterozygous germline mutations of the PHOX2B gene occur in pedigrees and are associated with neuroblastoma, ganglioneuroblastoma, congenital central hypoventilation syndrome, and Hirschsprung disease. The human PHOX2B gene maps to chromosome 4p13 and consists of three exons encoding a highly conserved 314–amino acid protein with two polyalanine repeats of 9 and 20 residues C-terminal to the homeodomain. Whereas the PHOX2B variants in congenital central hypoventilation syndrome largely involve expansions of the second polyalanine repeat within the C-terminus of the protein, those associated with neuroblastic tumors are nearly always frameshift and truncation mutations. More recently, whole-allele deletions resulting in the reduction of the protein have been reported. Proposed mechanisms contributing to neuroblastoma predisposition include partial loss of function with the preserved ability to suppress cellular proliferation but not to promote differentiation, complete loss of function due to functional haploinsufficiency, and both dominant-negative and gain-of function effects. Mutated PHOX2B also suppresses proliferation in vitro; heterozygous insertion of two frameshift variants, 931del5 and 693del8, into the mouse Phox2b locus resulted in impaired proliferation of sympathetic ganglion progenitors and biased differentiation toward the glial lineage. Aberrant Phox2b expression in the zebrafish models has been shown to cause an arrest in the normal maturation of sympathetic neurons, leading to immature cells that are resistant to retinoic acid-induced differentiation. Allelic deficiency of PHOX2B causes a decrease in the terminal differentiation markers th and dbh in sympathetic ganglion cells. The neuroblastoma-associated frameshift mutations 676delG and K155X—but not the R100L missense mutation—functioned dominant negatively to impede differentiation. Thus a reduced dosage of PHOX2B during development, through either a heterozygous deletion or dominant-negative mutation, imposes a block in the differentiation of sympathetic neuronal precursors, resulting in an immature cell population that is likely to be susceptible to secondary transforming events. The block in differentiation of immature sympathetic neurons caused by certain PHOX2B variants could be due to their inability to bind to interacting proteins critical to this process. The neuronal calcium sensor protein HPCAL1 (VILIP-3) exhibits strong binding to wild-type PHOX2B , but only weakly or not at all to neuroblastoma-associated frameshift and truncation variants leading to impaired subcellular localization of HPCAL1 and impaired differentiation in vitro.
Anaplastic Lymphoma Kinase Mutations
Dominant mutations in the anaplastic lymphoma kinase (ALK) tyrosine kinase receptor have been identified in approximately 50% of familial neuroblastoma cases. Several distinct germline mutations have been described to date—R1275Q, R1192P, T1151R, and G1128A—with R1275Q being the most frequent ( Table 54-1 ). ALK germline mutations generally occur in the context of multifocal tumors, although in one study, affected patients did not necessarily have younger age of onset. Further guidelines on screening need to be derived from a consensus analysis of inherited cases, but it would seem that in addition to a family history, the presence of multifocal tumors would call for genetic screening. In fact, in the United States, genetic testing for ALK and PHOX2B mutations is recommended for every newly diagnosed patient with a family history of neuroblastic tumors.
| Mutation | Tumor/Cell Line | Effect of Mutation | ALK Domain | Reference(s) |
|---|---|---|---|---|
| ALKΔ2-3 | T/C | GOF, can occur in amplified ALK | Δ224-318 | |
| ALKΔ4-11 | T/C | GOF, can occur in amplified ALK | Δ318-782 | |
| K1062M | T/C | GOF | JMR | |
| G1128A * | T | GOF | P-loop | |
| M1166R * | T | Ligand dependent or GOF | αC helix | |
| I1171N * | T | GOF | αC helix | |
| F1174I * | T | GOF | αC helix | |
| F1174L/S * | T/C | GOF | αC helix | |
| R1192P * | T | GOF | β4 strand | |
| F1245C * | T | GOF | C-loop | |
| R1275Q * | T/C | GOF | A-loop | |
| T1087I | T | Ligand dependent | JMR | |
| D1091N | T/C | Ligand dependent | β1 strand | |
| A1099T | T | Ligand dependent | β2 strand | |
| T1151M * | T | Ligand dependent | β3 strand | |
| A1234T * | T | Ligand dependent | αE helix | |
| 1464STOP | T | Ligand dependent | C-terminal to kinase domain | |
| I1250T * | T | Kinase dead | C-loop | |
| R1061Q | T | Unknown | JMR | |
| T1151R * | T | Unknown | β3 strand | |
| I1170T/S * | T | Unknown | αC helix | |
| F1174C/V * | T | Unknown | αC helix | |
| R1231Q * | T | Unknown | αE helix | |
| L1240V * | T | Unknown | αE helix | |
| F1245I/L/V * | T/C | Unknown | C-loop | |
| R1275L * | T | Unknown | A-loop | |
| Y1278S * | T | Unknown | A-loop |
Germline Susceptibility Variants
Genome-wide association studies have identified SNPs within or adjacent to genes that predispose to sporadic neuroblastoma: BARD1 (BRCA1-associated RING domain 1), LINC00340 (long intergenic nonprotein coding RNA 340, CASC15 ), LMO1 (LIM domain only 1), DUSP12 (dual-specificity phosphatase 12), DDX4/IL31RA (DEAD box polypeptide 4/interleukin [IL]31 receptor A), HSD17B12 (hydroxysteroid [17-beta] dehydrogenase 12), LIN28B (lin-28 homolog B), and HACE1 (HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase–1). The BARD1 SNP effect at chromosome 2q35 has been confirmed in African-American children, and the BARD1β isoform and LIN28B are oncogenic in neuroblastoma. Patients with neuroblastoma who were homozygous for the risk alleles were more likely to have metastatic disease, amplification of MYCN (v-myc avian myelocytomatosis viral oncogene neuroblastoma-derived homolog), and disease relapse. However, the absolute risk conferred by the susceptibility allele is extremely small, and the risk that neuroblastoma may recur in families with these risk alleles is estimated to be very low. A common copy number variation at 1q21.1 was also associated with neuroblastoma, the significance of which is uncertain. The mechanisms through which these risk alleles and associated genes contribute to neuroblastoma initiation and/or maintenance require further investigation.
Genetic Aberrations in Sporadic Neuroblastoma
DNA Ploidy
Neuroblastomas are classified into those with diploid modal chromosomal content (with a deoxyribonucleic acid [DNA] index of 1) or hyperdiploid modal chromosomal content (DNA index greater than 1). Hyperdiploidy is associated with whole chromosome gains and overall favorable biology, whereas diploidy is associated with segmental chromosomal rearrangements and unbalanced translocations. Such segmental chromosomal aberrations seem to affect certain chromosomes—1p, 11q, and 17q—although the biologic significance is unclear at present. Hyperdiploid tumors are often localized and have a favorable outlook, whereas diploid tumors not only tend to have a poor prognosis but also are associated with other high-risk genetic aberrations and MYCN amplification. Originally, infants (those younger than 1 year) with a hyperdiploid modal chromosome number were found to respond well to conventional therapy, whereas those with advanced stage disease with diploidy failed to respond to conventional therapy. Subsequent studies have shown that tumor cell ploidy predicts responsiveness to therapy in children up to 18 months of age with advanced stage neuroblastoma without MYCN amplification.
MYCN Amplification
MYCN, the first oncogene proven to be of clinical significance in neuroblastoma and gene amplification, occurs in approximately 20% to 30% of primary tumors and 90% of cell lines. MYCN amplification is generally associated with advanced disease, poor outcome, and rapid tumor progression in neuroblastoma. It occurs in 5% to 10% of early stage tumors and in half of the tumors of patients with advanced stage disease. The detection of MYCN gene amplification in tumor cells at diagnosis is used by most cooperative groups to stratify patients to more intensive therapy. MYCN copy number appears to be consistent throughout the natural course of the disease, that is, at diagnosis and relapse and between primary and metastatic tumors in individual patients. The Children’s Oncology Group (COG) currently classifies MYCN amplification as more than 10 copies per diploid cell; it is unclear whether lower levels of gain (i.e., MYCN copy number between 3 and 10) confer an adverse outcome. A correlation usually exists between amplification and increased MYCN expression, and tumors with MYCN amplification generally express higher levels of MYCN than do nonamplified tumors; however, it has been reported that high expression is not linked to outcome. MYC deregulation is common in high-risk tumors without MYCN amplification, suggesting that MYC signaling could contribute to the high-risk phenotype.
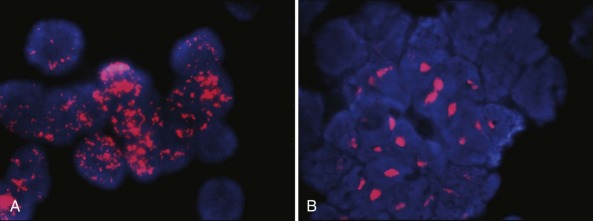
Amplification manifests cytogenetically as double minutes or homogeneously staining regions; the latter occur mainly in tumors and the former in cell lines ( Fig. 54-2 ). However, there is no difference in clinical outcome whether MYCN amplification is manifested as double minutes or homogeneously staining regions in tumor samples obtained at diagnosis. The genomic region amplified with MYCN is around 500 to 1000 kb, and several genes have been shown to be coamplified with MYCN in neuroblastoma. These genes include DDX1 (DEAD box helicase 1) , NAG ( NBAS [neuroblastoma amplified sequence]), and NCYM (MYCNOS—MYCN opposite strand), and it is possible that coamplification of these genes may contribute to the tumor phenotype.
Role of MYCN during Development.
MYCN is a highly conserved bHLH leucine zipper transcription factor located on chromosome 2p24.3 and a member of the myc family of oncogenes. The coding regions of both MYC and MYCN are highly homologous, with each having long 5′ and 3′ untranslated regions and gene products of similar sizes. Both dimerize with Max and bind DNA at consensus E-box sequences (CANNTG; reviewed in reference ). Mice homozygous for disrupted MYCN die at around 10 days of gestation and have multiple organ defects, including tissues of the central and peripheral nervous systems, mesonephros, lung, gut, and heart.
MYCN is highly expressed in fetal and neonatal development, specifically in the forebrain, hindbrain, and kidney, but not in adult tissues. MYCN is expressed in the fetal brain up to the onset of differentiation and is also present at high levels in neuroblasts migrating from the neural crest through the adrenal cortex. MYCN regulates overlapping stem-related gene expression programs in neuroblastoma and neural stem cells. MYCN induces a stemlike state by blocking differentiation pathways and activating self-renewal and pluripotency factors such as KLF2 and KLF4 (Kruppel-like factor 2 and 4) and LIN28B.
Role of MYCN in Tumorigenesis.
Suppression of MYCN expression in neuroblastoma cell lines results in a more differentiated phenotype. Indeed downregulation of MYCN occurs in neuroblastoma cells induced to differentiate with chemical agents, and MYCN overexpression can block retinoic acid–induced differentiation. Deletion of MYCN in neural progenitor cells showed decreased brain size and an increase in neuronal differentiation. Enhanced expression of MYCN and the mutant H- ras gene cause tumorigenic conversion of primary rat embryo fibroblasts, and these transformed cells elicit tumors in athymic mice and isogenic rats. Targeted expression of MYCN to the neuroectoderm of mice using a tyrosine hydroxylase promoter resulted in neuroblastoma tumors with chromosomal copy number abnormalities syntenic to those found in human tumors. The prolonged latency together with the additional chromosomal anomalies in these tumors suggests that additional genetic mutations are required for neuroblastoma formation.
MYCN is involved in all aspects of tumor cell metastasis: adhesion, motility, and invasion (reviewed in reference ). It downregulates integrins α1 and β1 to allow invasion. MYCN-amplified tumors have increased angiogenesis: overexpression of MYCN is associated with proangiogenic factors such as angiogenin and vascular endothelial growth factor (VEGF) through the PI3K/mTOR (mammalian target of rapamycin) pathway.
Role of MYCN in Altered Tumor Metabolism.
Similar to other cancers, neuroblastomas require abnormally high amounts of glucose and other macromolecules to enable their continued unchecked proliferation. These needs are met through alterations in key metabolic pathways that can potentially be harnessed for the development of new anticancer therapies.
Normal cells generate adenosine triphosphate (ATP) from glucose through oxidative phosphorylation when oxygen is abundant. During oxygen deprivation, which occurs commonly in solid tumors, including neuroblastoma, ATP generation occurs through glycolysis (the Warburg effect). Genes involved in glycolysis are selectively activated by hypoxia-inducible factor–1α (HIF1a), which is upregulated in hypoxic states. HIF1a is preferentially expressed in MYCN-amplified neuroblastoma cells; moreover, high levels of MYCN have been shown to override HIF1a inhibitory effects on cell cycle progression, thus enabling continued proliferation under hypoxic conditions. Therefore both deregulated MYCN and HIF1a cooperate to contribute to the Warburg effect in neuroblastoma cells. Moreover, expression of glycolytic genes phosphoglycerate kinase 1 (PGK1) , hexokinase 2 (HK2) , and lactate dehydrogenase A (LDHA) were higher in MYCN-amplified neuroblastoma than in those without MYCN amplification, with depletion of LDHA inhibiting tumorigenesis in vivo, suggesting that these metabolic pathways could be targeted therapeutically.
MYCN-amplified cells have also been shown to depend on exogenous glutamine for their survival. MYCN-amplified neuroblastoma cells overexpress high-affinity glutamine transporters and other glutaminolytic enzymes that correlate with poor prognosis. Glutamine depletion in MYCN-amplified neuroblastoma cells led to apoptosis through p53-independent PUMA (p53-upregulated modulator of apoptosis) and NOXA induction, an effect that is mediated by activation of the activating transcription factor–4 (ATF4). ATF4 has both context-dependent prosurvival and proapoptotic effects. In MYCN-amplified cells, it appears to have a prodeath role, because ATF4 agonists and glutaminolysis inhibitors potently induced apoptosis in vitro and inhibited tumor growth in vivo, suggesting that ATF4 agonists could be potential therapeutics for MYCN-amplified neuroblastomas.
MYCN in Immune Surveillance.
MYCN is also important in immune surveillance in that it represses several chemoattractant proteins that are required for recruitment of natural killer T cells (NKTs). NKTs are important for producing proinflammatory cytokines to recruit immune cells, stimulate the maturation of dendritic cells, and generate antigen-specific T cells to target the tumor. Patients with MYCN-amplified neuroblastoma with bone marrow metastases had fewer bone marrow NKTs. One such protein repressed by MYCN is monocyte chemoattractant protein–1/chemokine (C-C motif) ligand 2 (MCP-1/CCL2). Deletion of MYCN in MYCN-amplified cells rescues MCP-1 production and NKT recruitment. Therefore avenues to improve NKT function are being explored as therapeutic modalities in neuroblastoma.
ALK Amplification and Mutation
The identification of somatic mutations in the ALK receptor in neuroblastoma represents the first therapeutically targetable genetic aberration in this disease. ALK, a cell surface tyrosine kinase receptor of the insulin receptor superfamily, is located in humans at chromosome 2p23 and contains an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain (reviewed in reference ). Ligand binding leads to receptor dimerization and activation via trans-autophosphorylation of tyrosine residues. The 177-kDa polypeptide encoded by the human ALK gene undergoes posttranslational modification, such as N-glycosylation, to generate a mature protein doublet of 220 and 190 kDa. ALK is preferentially expressed in the central and peripheral nervous systems during development (i.e., thalamus, hypothalamus, midbrain, cranial ganglia and olfactory bulb, and dorsal root ganglia). ALK levels decrease postnatally, so that in adults ALK expression is restricted to rare scattered neural cells, endothelial cells, and pericytes in the brain. The physiologic function of ALK is not clear, although its predominant expression in the nervous system during development suggests that it likely plays an important role in the development of the nervous system. Homozygous ALK-null mice are viable and develop without obvious anatomic abnormalities and a normal life span, other than increased basal dopaminergic signaling within the prefrontal cortex and an age-dependent increase in basal hippocampal progenitor proliferation, as well as mild behavioral phenotypes. In Drosophila melanogaster , the receptor is important for the development of visceral gut musculature because absence of dALK leads to the loss of mesodermal founder cells responsible for gut development. The zebrafish alk ortholog is expressed at high levels in the developing nervous system. Ectopic expression of alk during early neurogenesis results in increased cell proliferation and aberrant neurogenesis in the central nervous system (CNS), leading to mispositioning of differentiated neurons and activation of the MEK/ERK pathway. Morpholino-mediated and pharmacologic depletion of alk, however, did not affect neuron progenitor formation, but it did cause compromised neuronal differentiation and survival in the CNS.
Signaling by the Wild-Type ALK Receptor.
Pleotropin (PTN) and midkine (MK) have been reported as candidate ligands of mammalian ALK. Whether PTN and MK are true ligands of ALK remains controversial because some groups have failed to reproduce the binding and activation of ALK by PTN. Signaling by these two molecules may trigger dimerization and phosphorylation of ALK, leading to activation of downstream signaling pathways. In D. melanogaster and Caenorhabditis elegans , the ALK ligands are Jelly belly (Jeb) and hesitation behavior 1 (HEN1), respectively, with Jeb being structurally different from either PTN or MK. Ligand activation of ALK has been implicated in the inhibition of apoptosis and induction of neuronal cell differentiation through the mitogen-activated protein kinase (MAPK) pathway. Signaling through the phospholipase Cγ (PLCγ) pathway by activated ALK, leading to transformation of NIH 3T3 cells, has also been reported.
ALK Mutations in Neuroblastoma.
In human tumors the most common mechanism of constitutive ALK activation involves chromosomal translocations that result in the generation of oncogenic ALK fusion genes, such as nucleophosmin (NPM1)-ALK in anaplastic large cell lymphoma and echinoderm microtubule-associated protein like 4 (EML4)-ALK in non–small cell lung cancers (NSCLCs), among others. ALK translocations have not yet been reported in neuroblastomas, apparently because the ALK receptor is normally expressed by sympathetic neuroblasts, and a heterologous promoter does not need to be provided through translocation. Rather, ALK aberrations take the form of point mutations, amplification, and copy number gain. Activating mutations in ALK occur in 7% to 8% of sporadic neuroblastomas. ALK is also amplified in neuroblastoma, although always in combination with MYCN amplification, and amplified ALK appears to mediate overexpression alone in that the affected ALK allele does not harbor activating point mutations.
Altogether 12 different residues are known to be affected by ALK mutations ( Table 54-1 ). Mutations range from silent to activating, with the two major mutational hot spots being R1275 and F1174; the R1275Q mutation is found in both familial and sporadic neuroblastoma, whereas the F1174L mutation is restricted to sporadic tumors, presumably because it is embryonic lethal. Activating ALK mutations are associated with constitutive phosphorylation of ALK and that of downstream targets such as ERK, STAT3, and AKT. These ALK variants are for the most part retained intracellularly in the endoplasmic reticulum and Golgi and exhibit impaired maturation with defective N-linked glycosylation. Moreover, the constitutive activity of these variants does not require receptor dimerization. Their oncogenic potential has been demonstrated by cytokine-independent growth, transformation of NIH-3T3 fibroblasts in soft agar colony formation assays, and tumor formation in nude mice. Knockdown of ALK in ALK-mutant neuroblastoma cell lines led to significant inhibition of cell proliferation and induction of cell death, further emphasizing the strong oncogenic “addiction” of these cells to mutationally activated ALK.
Elucidation of the crystal structure of ALK in its inactive conformation and mapping of neuroblastoma-associated mutations has shown that many of the ALK residues that become mutated in neuroblastoma play structural roles in autoinhibition of ALK. Mutations allow unrestricted mobility of the alphaC helix, phosphate-binding loop, and N-terminal lobe, leading to kinase activation. Both the F1174L and R1275Q mutants show accelerated catalytic efficiency compared with wild-type ALK, with R1275Q exhibiting approximately fourfold increased catalytic efficiency and F1174L exhibiting approximately eightfold increased catalytic efficiency compared with the wild-type enzyme. This increased catalytic efficiency is at least partly due to the enhanced ATP binding affinity of these mutations for both ATP and peptide substrate.
The ALK mutants show varying degrees of transformation capabilities. ALK F1174L is considered the most aggressive of all ALK mutations in neuroblastoma, possessing higher transforming potential and kinase activity. Importantly, ALK F1174L also arises secondarily as a mechanism of resistance after an initial response to crizotinib in patients with ALK -rearranged cancers. Moreover, the F1174L mutation cosegregates with MYCN amplification in patients with neuroblastoma, and this combination is associated with a particularly poor prognosis. ALK F1174L potentiates the oncogenic activity of MYCN in vitro and in vivo, although by itself, ALK F1174L appears to be insufficient to cause the malignant transformation of neural crest–derived stem cells. When crossed with MYCN transgenic mice, mice overexpressing ALK F1174L in neural crest–derived cells gave rise to doubly transgenic progeny that developed aggressive neuroblastoma with 100% penetrance and a very short latency period. ALK F1174L /MYCN tumors exhibited increased MYCN dosage due to ALK F1174L -induced activation of the PI3K/AKT/mTOR and MAPK pathways, coupled with suppression of MYCN pro-apoptotic effects. Similarly, coexpression of ALK F1174L and MYCN in zebrafish markedly increased the frequency of tumor formation and accelerated the time of onset. MYCN overexpression induces adrenal sympathetic neuroblast hyperplasia, blocks chromaffin cell differentiation, and ultimately triggers a developmentally timed apoptotic response in the hyperplastic sympathoadrenal cells. Coexpression of activated ALK with MYCN provides prosurvival signals that block this apoptotic response and allow continued expansion and oncogenic transformation of hyperplastic neuroblasts, thus promoting progression to neuroblastoma.
ALK mutations show no predilection for particular clinical stages of tumor progression. With the exception of F1174L mutations that occur preferentially in MYCN-amplified tumors, somatic ALK mutations do not appear to be associated with survival, although a more recent study reported that mutation-positive cases had a decreased overall survival (OS) probability. However, ALK overexpression in the absence of mutation or amplification does appear to be associated with OS. ALK RNA and protein overexpression is detectable in approximately 90% of primary tumors and in some reports is associated with activation of downstream signaling. The significance of this phenomenon, as well as the mechanism underlying activation of ALK, is presently unclear. Passoni et al. (2009) argue that it is the level of expression that dictates oncogenicity, based on the fact that constitutive phosphorylation of ALK was observed in cells with high expression, whereas those with low expression lacked activation. This observation, plus the fact that genetic knockdown of wild-type ALK caused a decrease in cell proliferation, suggest that overexpression of wild-type ALK may have some degree of proproliferative activity. Other studies have shown that neuroblastoma with elevated expression of wild-type ALK had clinical and molecular phenotypes similar to those of tumors with mutated ALK gene expression. These data suggest that mechanisms other than mutation and amplification can lead to ALK activation.
ALK Amplification.
ALK is amplified in around 2% to 3% of primary neuroblastoma cases. Amplification tends to coexist with MYCN amplification; in fact, ALK amplification has been detected in up to 15% of primary neuroblastomas with MYCN amplification. ALK amplification does not appear to have any independent prognostic value in neuroblastoma. ALK copy number gain appears to be more common than mutation or amplification, occurring in 15% to 20% of cases, and usually involves whole chromosome 2 gain. Such chromosome 2p gains are associated with significantly increased ALK expression, correlating with poor survival.
ALK Signaling in Neuroblastoma.
Constitutively activated ALK as a result of ALK amplification binds to the activated Src homology 2 domain–containing adaptor protein ShcC and thereby activates downstream signaling. Inhibition of this binding impairs survival, differentiation, and motility of neuroblastoma cells through blockage of the MAPK and PI3K/AKT pathways and impairs the induction of apoptosis. Gain-of-function ALK mutations also commonly signal through the MAPK, PI3K/AKT/mTOR, and STAT pathways to regulate cell growth, transformation, and antiapoptotic signaling (reviewed in references and ).
ATRX Mutations
The alpha thalassemia/mental retardation syndrome X-linked (ATRX) gene is mutated primarily in 44% of neuroblastoma tumors in adolescents and young adults, which largely have an indolent tumor phenotype compared with that in children and infants. These loss-of-function mutations are associated with absence of the ATRX protein in the nucleus and telomere lengthening, the mechanism of which is unclear. ATRX is involved in the regulation of ATP-dependent chromatin remodeling, nucleosome assembly, and telomere maintenance. ATRX also plays a role in epigenetic regulation of gene expression by controlling the deposition of histone H3.3 at transcriptionally silent regions of the genome, which may potentially lead to increased expression of oncogenes in tumors with ATRX mutations. ATRX mutations correlate with older age at diagnosis (age >5 years) and chronic or indolent disease. The ATRX gene is present on the X chromosome, and both males and females were affected. These mutations are not thought to be associated with MYCN amplification, although further studies are required to validate this supposition.
Chromosomal Abnormalities in Neuroblastoma
Chromothripsis
Chromothripsis is a phenomenon that has been recently identified in 18% of high-stage neuroblastomas. Chromothripsis is a local shredding of chromosomes with subsequent random reassembly of the fragments. These structural alterations recurrently affected ODZ3 ( TENM1 [teneurin transmembrane protein 1]), PTPRD (protein tyrosine phosphatase, receptor type, D), and CSMD1 (CUB and Sushi multiple domains 1) genes involved in neuronal growth cone stabilization. ODZ3 and PTPRD encode transmembrane receptors expressed in the developing nervous system localizing to axon and axonal growth cones. Chromothripsis-related structural aberrations were associated with amplification of MYCN or cyclin-dependent kinase–4 (CDK4) and loss of heterozygosity (LOH) of chromosome 1p.
1p Loss of Heterozygosity
LOH of the short arm of chromosome 1p is a common occurrence in neuroblastoma. The common region of LOH is within 1p36.2-1p36.3. However, this region may be much larger, extending from 1p35 to the terminal end. This abnormality has been reported not only in neuroblastoma but also in melanoma, pheochromocytoma, and medullary thyroid carcinoma, all of which are neural crest–derived tumors. LOH of 1p occurs in 30% to 40% of neuroblastomas and is positively correlated with older age, advanced stage, MYCN amplification, and a poor outcome. When MYCN amplification and 1p LOH are present together, they define a genetically distinct and very aggressive subset of neuroblastoma. Because cases with MYCN amplification represent a subset of patients with 1p deletion (because MYCN amplification is rarely found in the absence of 1p deletion), it is believed that 1p deletion may precede the development of MYCN amplification. Although 1p loss is also present in almost all samples with MYCN amplification, it is also found in high-risk cases without MYCN amplification.
Therefore it follows that loss or inactivation of a gene or genes on 1p would be critical for the development or progression of neuroblastoma. This scenario has been suggested by reports of constitutional 1p36 alterations in patients with neuroblastoma. Transfection of chromosome 1p into a neuroblastoma cell line restores a differentiated phenotype and abrogates tumorigenicity. In addition, cell fusion experiments between MYCN amplified and single copy cells have resulted in abrogation of MYCN expression, suggesting than MYCN could be regulated by a gene located on 1p. Several candidate tumor suppressor genes have been identified, including CHD5 (chromodomain helicase DNA binding protein 5) , KIF1B (kinesin family member 1B), TP73 (tumor protein p73), MIR34A (microRNA 34A), CAMTA1 (calmodulin binding transcription activator) , and CASZ1 (castor zinc finger 1).
CHD5.
CHD5, a member of the CHD family of proteins involved in chromatin remodeling, is preferentially expressed in the nervous system and maps to a small region of deletion on 1p36.3 in neuroblastomas. Expression of CHD5 has been found to be low or absent in cell lines and tumors with 1p deletion. Low expression of CHD5 is also highly correlated with MYCN amplification, advanced stage, and unfavorable histology. When CHD5 was expressed in neuroblastoma cell lines with low or absent expression, clonogenicity and tumor growth were abrogated. Mouse models with loss of a region corresponding to human 1p36 implicated CHD5 as a tumor suppressor that controls proliferation, apoptosis, and senescence via the p19 Arf /p53 pathway. CHD5 mutations, however, are rare in neuroblastoma, although it is now becoming clear that complete inactivation of the gene does not appear to be necessary for malignant transformation. Rather, gene dosage may be another mechanism that regulates tumor suppressor activity as shown for some PHOX2B mutations. In a study by Bagchi et al., (2007) increased dosage of CHD5 (as in wild-type or three copies) led to tumor-suppressive properties, whereas decreased dosage (as in heterozygous 1p deletion) enhanced immortalization, spontaneous foci formation, and sensitivity to oncogenic transformation. In addition, it has been suggested that almost complete inactivation of the second allele may occur by an epigenetic phenomenon because the CHD5 promoter was found to be highly methylated in two cell lines that lacked CHD5 expression.
KIF1B.
The kinesin family member, KIF1B, is located on 1p36.2 and is thought to function as a tumor suppressor gene in neuroblastoma and pheochromocytoma. Germline loss-of-function mutations have been detected in patients with neuroblastoma, leading to abrogation of the apoptosis that is a requisite to normal neuronal developmental culling when nerve growth factor (NGF) becomes limiting. Neuroblastoma cells have been shown to undergo apoptosis when NGF is withdrawn, which is mediated through the EglN3 prolyl hydroxylase. KIF1B acts downstream of EglN3. Similar to the case with CHD5, KIF1B haploinsufficiency may be sufficient for loss of its tumor suppressor activity, especially if combined with loss or abnormalities of other genes on 1p36 such as CHD5. In support of this theory, when KIF1B levels were decreased to 50% using short hairpin (sh)RNA knockdown, inhibition of apoptosis was observed.
CASZ1.
Deletion of CASZ1, a neuronal differentiation gene, has been implicated in neuroblastoma tumorigenesis. CASZ1 is the human homologue of the Drosophila zinc finger transcription factor, castor. Low expression of CASZ1 via LOH or epigenetic suppression is associated with poor prognosis in patients with neuroblastoma. Moreover, the restoration of CASZ1 in neuroblastoma cells suppresses cell proliferation by activation of pRB in G1 and inhibition of the G2/M regulators cyclin B1 and Chk1 and induces cell differentiation. Decreased CASZ1 expression in neuroblastoma is thought to be due to upregulation of one of the three core subunits of the polycomb repressor complex 2, EZH2 (enhancer of zeste 2). This methyltransferase regulates trimethylation of histone H3 on lysine 27 (H3k27me3 [histone H3 trimethyl lysine 27]), which is associated with gene silencing. Analysis of expression data sets revealed that patients with neuroblastoma who have a poor prognosis have increased levels of EZH2 expression. Silencing of EZH2 leads to decreased H3K27me3 and increased expression of CASZ1. Thus a model has been suggested in which one allele of CASZ1 could be lost by 1p LOH, with the remaining allele epigenetically silenced by EZH2-mediated H3k27me3. Moreover, EZH2 also was found to silence a number of tumor suppressors that control differentiation in neuroblastoma, such as clusterin (CLU) , runt-related transcription factor–3 (RUNX3) , and nerve growth factor receptor (NGFR) . Genetic and pharmacologic inhibition of EZH2 inhibited neuroblastoma cell growth and induced differentiation.
MIR34A .
The microRNA (miRNA)-34a (MIR34A) on 1p36.23 is expressed at lower levels in primary tumors and cell lines with 1p deletion and is also thought to function as a tumor suppressor gene. Reintroduction of this miRNA into neuroblastoma cell lines with 1p deletion causes a dramatic reduction in cell proliferation through the induction of a caspase-dependent apoptotic pathway and by reducing levels of E2F3, a transcriptional inducer of cell-cycle progression. MIR34A also increases during retinoic acid–induced differentiation of neuroblastoma cells. Indeed, targeted delivery of miRNA-34a using nanoparticles conjugated to an anti-GD2 antibody that is expressed on the surface of virtually all neuroblastoma cells was shown to result in decreased tumor growth in orthotopic xenograft models, highlighting its therapeutic potential. In addition, MIR34A has also been found to directly target and downregulate MYCN and B-cell lymphoma–2 (BCL2). MIR34A causes significant suppression of cell growth through increased apoptosis and decreased DNA synthesis in neuroblastoma cell lines with MYCN amplification.
ARID1 Mutations.
Deletions and sequence alterations in the chromatin-remodeling genes ARID1A and ARID1B (AT-rich interactive domain 1A and 1B) have been reported in 11% of tumors. These mutations were associated with early treatment failure and decreased survival. ARID1 family genes are integral components of the switch/sucrose nonfermentable (SWI/SNF) neural progenitor-specific chromatin-remodeling BRG1-associated factor (BAF) complex that is essential for the self-renewal of multipotent neural stem cells. ARID1A was affected by insertional and point mutations predicted to result in premature termination of the protein and deletion of the other allele at 1p36, largely leading to biallelic inactivation. Hemizygous deletions encompassing the whole coding sequence, intragenic hemizygous deletions, and splice-site and missense mutations targeting ARID1B were also reported in this study. High expression of members in the neural progenitor BAF complex correlated with a high-risk neuroblastoma phenotype, whereas high expression of components of the neuron-specific BAF complex or downstream neuritogenesis target genes correlated with low-risk neuroblastoma. Thus it is possible that disrupted BAF complex signaling preserves an undifferentiated progenitor state, although this preservation has not as yet been demonstrated.
LIN28B Overexpression
Overexpression of the LIN28B gene that encodes a protein that binds small RNAs has been reported in neuroblastoma. Lin28b is highly expressed in stem cells and developing tissues and regulates germ cell development, skeletal myogenesis and neurogenesis, and glucose metabolism. It is a master regulator of pluripotency in embryonic stem (ES) cells, and in combination with NANOG (Nanog homeobox), OCT4 (organic cation transporter–4), and SOX2 (sex-determining region Y box–2), it can reprogram differentiated cells to pluripotent stem cells. High LIN28B expression was found to be an independent risk factor for adverse outcome in persons with neuroblastoma. In neuroblastoma cells, LIN28B, through repression of the let-7 miRNA family, caused elevated MYCN expression and inhibition of normal neuroblast and neuroblastoma differentiation, whereas overexpression of LIN28B in nonmalignant neuroblasts drives proliferation. Targeted expression of LIN28B in the sympathetic adrenergic lineage induced development of neuroblastomas marked by low let-7 miRNA levels and high MYCN protein expression. Mechanisms underlying the overexpression of LIN28B in neuroblastoma are currently unclear. LIN28B has also been identified in a genome-wide association study of patients with neuroblastoma as influencing susceptibility to neuroblastoma, and high expression was associated with worse OS.
Chromosome 17q Gain
Gain of chromosome 17q is the most common cytogenetic abnormality in neuroblastoma; it occurs in more than 60% of neuroblastomas and is associated with an unfavorable prognosis and metastatic disease. Gain usually takes the form of one to three extra copies. The break points vary, but in general, gain of a region from 17q22-qter is observed. Partial gain often results from unbalanced translocation of 17q21-25 to another chromosome. Unbalanced 1;17 translocations occur in primary neuroblastoma and result in loss of distal 1p with gain of 17q material. Unbalanced 17q gain is associated with MYCN amplification and most likely harbors an oncogene that contributes to neuroblastoma tumorigenesis. However, 17q translocation break points are not uniform and can involve other partner chromosomes, especially 11q. The break point positions on 11q were found to be variable, whereas all break points on 17q appeared to cluster proximal to position 43.1 Mb on the DNA sequence map.
Isogenic cell lines derived from MYCN-driven murine tumors in transgenic mice showed gains of regions syntenic with human 17q. One of the candidate genes on 17q is survivin, the expression of which is significantly associated with a poor prognosis. Other candidate genes in the 17q region are nm23-H1 and nm23-H2, which are both targets of MYCN. Nm23-H1 binds to Cdc42, which is encoded on 1p36 and prevents neuroblastoma cell differentiation. Overexpression of Nm23 due to gain of 17q and induction by MYCN combined with decreased expression of Cdc42 due to loss of 1p36 can block neuroblastoma tumor differentiation.
Chromosome 11q LOH
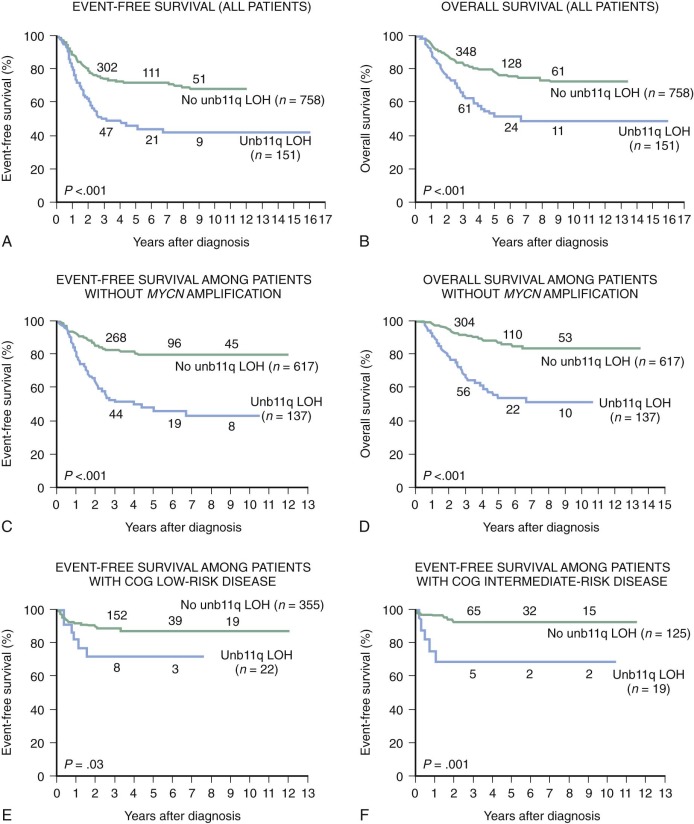
Allelic loss of 11q occurs in 35% to 45% of primary tumors. Chromosome transfer experiments involving transfer of an intact chromosome 11 into a neuroblastoma cell line induces differentiation. The common region of deletion has been mapped to 11q23, indicating that this could be a location for a neuroblastoma suppressor gene. Chromosome 11q loss is associated with multiple chromosomal breaks, possibly reflecting an underlying DNA repair defect. Chromosome 11q LOH occurs mainly in tumors without MYCN amplification and identifies a high-risk subset of patients with advanced stage, older age, and disease with unfavorable histologic features. Unbalanced deletion of 11q occurs in 15% to 20% of cases and is associated with a poor event-free survival (EFS) in patients with otherwise low-risk and intermediate-risk disease ( Fig. 54-3 ).
Chromosome 14q LOH
Deletion of 14q has also been reported in 25% of neuroblastomas, with a common region of deletion within 14q23-qter. LOH for 14q is inversely related to 1p36 LOH, MYCN amplification, and 11q LOH.
Other areas of chromosomal gain noted especially in tumors with an aggressive clinical phenotype, but not MYCN amplification, include gain of 1q and 12q. Chromosomal regions that have been deleted are 3p, 4p, 9p, and 18q, but these regions are all less common.
Neurotrophin Receptors in Neuroblastoma
Tropomyosin receptor kinase (TRK) receptors, when activated by their respective neurotrophins, mediate the survival and differentiation of neurons during development (reviewed in reference ). NGF-induced dimerization of TRKA (NTRK1) or brain-derived neutrotrophic factor (BDNF)–induced dimerization of TRKB receptors leads to activation of the tyrosine kinase domain, which facilitates binding of SHC and phosphotyrosine binding (PTB) domain protein adaptors and PLCγ protein binding, respectively, leading to activation of the major growth factor–regulated signaling pathways—the Ras/MAPK pathway and PI3K/3-phosphoinositide–dependent protein kinase–1 (PDK1)/AKT pathway—and to increases in Ca++ release and activation of the PLCγ pathway. The main ligand for TRKA, NGF, promotes survival and induces differentiation in developing sympathetic neuroblasts. Neuroblastoma tumor cells with high levels of TRKA expression differentiate in the presence of NGF in vitro but will undergo apoptosis in its absence. Depending on the tumor microenvironment, therefore, TRKA signaling could induce differentiation or regression of favorable neuroblastomas. During normal development, depletion of NGF occurs in sympathetic neurons, causing TRKA signaling to activate predetermined apoptotic pathways. It has been postulated that spontaneous regression of neuroblastomas is but a delay in this normal developmental pattern. A neurodevelopmentally regulated splice variant of TRKA, TrkAIII, has been identified that antagonizes the antioncogenic NGF/TRKA signaling and promotes neuroblastoma tumor growth. High levels of TRKA expression are associated with a good prognosis in neuroblastoma and are strongly correlated with favorable tumor stage, younger age, and nonamplified MYCN . Patients with hyperdiploid tumors with favorable outcome identified by mass screening were also shown to have very high TRKA expression.
The TRKB (NTRK2) transcript is expressed primarily in highly aggressive MYCN -amplified tumors. The ligand for TRKB is BDNF, and activation of TRKB by BDNF leads to enhanced proliferation, migration, angiogenesis and resistance to chemotherapy in neuroblastoma. Neuroblastoma cells that survive repeated exposures to cytotoxic agents express increasing levels of BDNF, suggesting that this pathway contributes to a multidrug-resistant phenotype. Moreover, hypoxia induces increased TrkB expression and may contribute to a phenomenon in the tumor microenvironment where autocrine activation of the BDNF/TrkB signaling enables survival of residual tumor cells after repeated rounds of chemotherapy. The full-length TRKB, which is expressed in about one third of tumors tested, is expressed primarily in those with MYCN amplification, whereas the truncated form resulting from alternative splicing, which lacks the tyrosine kinase domain, is expressed in ganglioneuromas and ganglioneuroblastoma. The truncated TRKB is thought to sequester BDNF and thus prevent TRKB signaling. Studies have shown that survival of neuroblastoma cells exposed to cytotoxic drugs is mediated partly by activation of the Trk receptor via PI3K, activation of Akt, and inactivation of glycogen synthase kinase–3 beta (GSK3B). Thus the response of neuroblastoma cells to chemotherapeutic agents depends on the levels of BDNF; tumor cells expressing low levels of TrkB exhibit minimal effects on exposure to cytotoxic agents in the setting of a BDNF-rich environment. BDNF/TrkB-induced increases in HIF1a may lead to increased VEGF and angiogenesis in neuroblastoma. Thus hypoxia-associated increases in neuroblastoma cell invasiveness can be blocked by inhibition of TrkB. Activation of the BDNF/TrkB signaling pathway stimulates tumor cell survival and angiogenesis and contributes to chemotherapy resistance and anoikis.
TRKC, like TRKA, is involved in the biology of favorable neuroblastomas, and expression corresponds to lower stages. Although its expression is found in 25% of primary neuroblastomas, it does not have independent prognostic significance, because all tumors with TRKC expression also have TRKA expression.
Apoptosis
In contrast to most human tumor types, the p53 gene is very rarely mutated in neuroblastoma at diagnosis but is found in chemotherapy resistance. Other mechanisms of p53 loss of function, such as cytoplasmic sequestration, mouse double minute 2 (MDM2 protooncogene) amplification, or TWIST-mediated suppression have been proposed, but their contribution appears to be limited. Although basal p53 expression in neuroblastoma cells is largely confined to the cytosol, p53 protein levels were found to increase mainly in the nucleus after radiation-induced DNA damage.
Epigenetic modification of the pro-apoptotic gene caspase-8 has been observed in neuroblastomas. Up to 70% of human neuroblastoma cell lines and 25% of primary tumors tested lack caspase-8 expression and fail to undergo apoptosis. Loss of expression of both caspase 8 (apoptosis-related cysteine peptidase, CASP8 ) alleles is correlated with methylation of the caspase-8 gene, and treatment with the demethylating agent decitabine restored caspase-8 expression and increased susceptibility to doxorubicin-induced apoptosis. In addition, methylation has been shown to be the mode of silencing of other genes involved in apoptosis: the four tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) apoptosis receptors and the caspase-8 inhibitor FLICE-like inhibitory protein (FLIP).
Overexpression of BCL2, an inhibitor of apoptosis, has been found to be associated with unfavorable histology and MYCN amplification. Neuroblastoma cells and tumors are primed for death with sequestration of BIM, a direct activator of apoptosis, either by the prosurvival BCL-2 or myeloid cell leukemia–1 (MCL-1) proteins. This pattern of survival dependency has been shown to predict the pattern of response to Bcl-2 antagonists. The Bcl-2/Bcl-xl/Bcl-w inhibitor ABT-737 was active against Bim : Bcl-2 primed tumor xenografts but not against cells where Bim was sequestered by Mcl-1. In isogenic cell lines at diagnosis and relapse, therapy resistance was shown not to be mediated by upregulation of Bcl-2 family members or loss of Bim priming but by repression of Bak and/or Bax-mediated apoptosis.
Therapy Resistance
The multidrug resistance gene encoding P-glycoprotein is thought to function by causing enhanced drug efflux from the cell, and its overexpression has been found to predict outcome of therapy for neuroblastoma. The multidrug resistance-associated protein (MRP), like P-glycoprotein, mediates resistance to a number of drugs. MRP is expressed by neuroblastoma tumors of all stages. Tumors with MYCN amplification have been shown to have significantly higher expression levels of MRP than do those with normal MYCN copy numbers. Reduced MRP expression levels correlate with differentiation of neuroblastoma cells in vitro. In addition, an association has been found between high levels of MRP expression and poor outcome, which is independent of MYCN amplification. It is possible that both multidrug resistance protein–1 (MDR1) and MRP function together with several other factors that confer resistance to drug therapy in neuroblastoma such as MYCN amplification, TRKB signaling, or loss of p53 expression.
MYCN-amplified tumors were found to possess a population of cells with CSC characteristics that was enriched during prolonged drug selection. Treatment with the histone deacetylase (HDAC) inhibitor vorinostat led to histone acetylation and increased the sensitivity of these cells to chemotherapy, with loss of their stemness properties and downregulation of stemlike marker genes.
Acquired Resistance to Targeted Therapies
The dissection of the cancer genome that has resulted in the identification of targetable oncogenic drivers has propelled personalized medicine protocols aimed at treating subsets of patients whose tumors harbor sensitizing mutations. Although pediatric tumors have a relatively silent mutational landscape compared with adult tumors, targeted agents are being used in several cancers, including neuroblastoma. These agents include small-molecule inhibitors of ALK, TRKB, aurora kinase, and PI3K/mTOR, to name a few. Targeted therapies can induce impressive clinical responses, but relapse due to acquired resistance is a major challenge. Several mechanisms lead to acquired resistance to targeted agents: acquisition of secondary mutations or amplification of the drug target itself, activation of alternative or downstream signaling pathways, and changes in drug efflux or bioavailability (reviewed in reference ). Secondary mutations in the target gene such as gatekeeper mutations preserve pathway activity and confer resistance by interfering with drug binding. Engagement of alternative receptor–mediated pathways has also been reported as causing resistance. Resistance to targeted therapy can also occur through nongenetic mechanisms, such as a reversible drug-tolerant state in a subpopulation of cells that is maintained by a chromatin modification, implicating an epigenetic basis of drug resistance. Another mechanism involves transdifferentiation or histologic transformation with epithelial to mesenchymal transition in the absence of any known genetic mechanism.
Tumor Angiogenesis
Neuroblastoma is characterized by prominent angiogenesis, and neuroblastoma cells have been shown to induce angiogenesis in the chick embryo chorioallantoic membrane assay. Increased tumor vascularity and microvascular proliferation is correlated with widely disseminated disease, MYCN amplification, unfavorable histology, and a poor outcome. Such aggressive tumors are associated with high expression of VEGF, basic fibroblastic growth factor, and platelet-derived growth factor A and integrins, which are markers of active angiogenesis. The presence of angiogenesis appears to be influenced by the cellular composition of the tumor in that in highly aggressive stroma-poor neuroblastoma, angiogenesis is present as a result of the secretion of angiogenic stimulators, whereas in stroma-rich tumors, numerous angiogenic inhibitors secreted by Schwann cells appear to maintain the inhibitory phenotype. The Schwann cells in neuroblastoma tumors have been shown to have very low tumor vascularity with production of angiogenesis inhibitors, such as tissue inhibitor of matrix metalloproteinase–2 (TIMP-2), pigment epithelium-derived factor, and SPARC (secreted protein acidic and rich in cysteine), a calcium-binding matricellular glycoprotein. SPARC expression has been found to be inversely correlated with the degree of malignant progression in neuroblastoma tumors, and neutralizing SPARC with antibodies reverses the antiangiogenic activity of Schwann cell–conditioned media.
Metastasis
Metalloproteinases such as matrix metalloproteinase–9 (MMP9), along with CD44 and NM23-H1, which regulate tumor cell adhesion and migration, may play a role in metastasis. Overexpression of MMP2 and MMP9 is associated with tumor invasion and metastasis in many types of cancer, whereas inhibitors of MMPs have been shown to suppress tumor invasion and angiogenesis. An association between increased levels of MMP2 and MMP9 and advanced-stage tumors has been observed in neuroblastoma. Caspase-8 has also been shown to be a metastasis suppressor gene. Decreased caspase-8 expression has been shown to occur during the establishment of neuroblastoma metastases in vivo, and reconstitution of caspase-8 expression in deficient neuroblastoma cells suppressed these metastases. Caspase-8 selectively potentiated apoptosis in metastasizing cells, and loss of caspase-8 allowed cellular survival in the stromal microenvironment and promoted metastases. In the TH-MYCN mouse model of neuroblastoma, caspase-8 was associated with increased metastasis, specifically to the bone marrow, possibly caused by upregulation of genes involved in epithelial-mesenchymal transition (EMT), decreased cell adhesion, and increased fibrosis.
Pathology
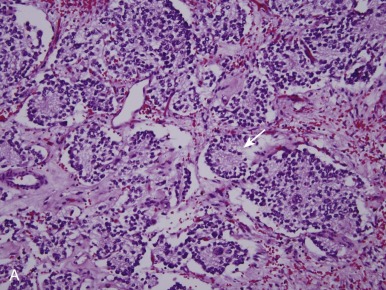
Neuroblastomas arise from primitive sympathetic precursors of the neural crest and belong to the family of small round blue cell tumors. The histopathology of the tumor cells correlates with stages of sympathetic nervous system development. Tumors are composed of small blue round cells that are uniformly sized and contain dense, hyperchromatic nuclei and scant cytoplasm. Surrounding the neuroblasts is stroma, known as Schwannian stroma. A typical feature of neuroblastoma is the presence of neuropil, which is made up of neuritic processes and is found in the majority of neuroblastomas. One of the pathognomonic features of neuroblastoma is the Homer-Wright pseudorosette, a collection of neuroblasts surrounding areas of neuropil, which occurs in up to half of cases (15% to 50%; Fig. 54-4, A ).
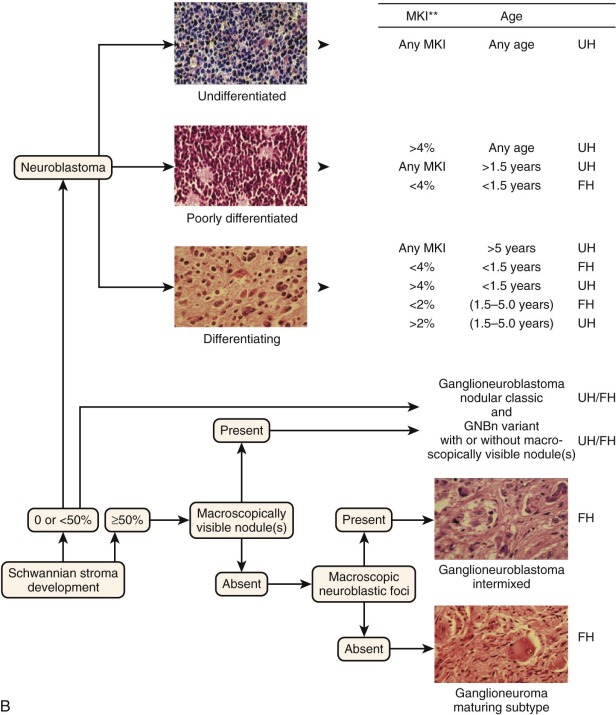
Neuroblastoma manifests as a spectrum of three histologic patterns ranging from neuroblastoma to ganglioneuroblastoma to ganglioneuroma, based on the degree of tumor cell differentiation. Neuroblastomas are composed of mostly small immature blue round cells with scanty cytoplasm, little evidence of differentiation, and high mitotic activity. Ganglioneuroblastomas are tumors with differentiated ganglion cells admixed with neuroblastic tissue. These tumors may vary from predominantly neuroblastic with rare ganglion cells to predominantly maturing cells with rare undifferentiated components, such as neuroblastic cells. If less than 50% of the cells are maturing cells, the tumor is termed a “maturing neuroblastoma,” and if more than 50% of the cells are maturing cells, it is termed a “ganglioneuroblastoma.” Ganglioneuroblastomas can also be focal or diffuse, and both types can exist within a single tumor. Two forms of ganglioneuroblastoma exist: (1) the intermixed variety, in which cells in various stages of differentiation are interspersed with small nests of neuroblasts, predicting a good outcome, and (2) the nodular type, in which hemorrhagic areas and macroscopic nodules are present, which is associated with a worse prognosis. Ganglioneuromas are fully differentiated tumors consisting entirely of maturing ganglion cells, neuropil Schwannian stroma, and nerve fibers.
Primary histologic diagnosis may be enabled by hematoxylin and eosin staining and light microscopy. Other techniques also help distinguish neuroblastomas from other small round blue cell tumors of childhood, such as immunohistochemistry with use of antibodies for neural markers, such as neurofilament protein, synaptophysin, neuron-specific enolase, ganglioside GD2, chromogranin A, and tyrosine hydroxylase. Electron microscopy studies may exhibit dense core-membrane–bound neurosecretory granules and microfilaments and parallel arrays of microtubules within the neuropil.
Tumor histology in neuroblastoma has traditionally been determined by the Shimada classification system. Tumors are classified as favorable or unfavorable based on three features: amount of stroma, degree of neuroblastic cell differentiation, and the mitosis-karyorrhexis index (MKI; the percentage of tumor cells in mitosis versus karyorrhexis). This system had one drawback in that it was age-linked, and age itself is a strong independent prognostic feature in neuroblastoma. The Joshi system was simpler in that it examined the presence of calcification and mitotic rate (less than or equal to 10 mitoses per 10 high-power fields) and was designed to be independent of age and stage; however, it did not have the same prognostic power as the Shimada system. Subsequently a unified classification, the International Neuroblastoma Pathology Classification (INPC), was established in 1999 and revised in 2003. This classification schema was formulated on the basis of the natural history of neuroblastoma of involution and maturation as described by Beckwith and Perrin (1963). In other words, it is based on the age-dependent normal ranges of morphologic features such as Schwannian stroma, degree of neuroblastic differentiation, and MKI and seeks to divide neuroblastic tumors into those with favorable and unfavorable histology ( Fig. 54-4, B ).
The INPC has four main morphologic categories ( Table 54-2 ).The first category is neuroblastoma (Schwannian-stroma poor), which is a tumor with nests of neuroblastic cells interspersed with little or minimal stroma. There are three subtypes, based on grade of differentiation: (a) undifferentiated, (b) poorly differentiated (some neuropil and <5% of cells exhibiting differentiation), and (c) differentiating (abundant neuropil and >5% of cells showing differentiation toward ganglion cells). The second category is ganglioneuroblastoma, intermixed (Schwannian stroma-rich), which is a tumor that contains well-defined microscopic nests of neuroblastic cells intermixed in ganglioneuromatous stroma. The nests are composed of neuroblastic cells in various stages of differentiation, but they are primarily composed of differentiating neuroblasts and maturing ganglion cells in a background of neuropil. The third category is ganglioneuroblastoma, nodular (composite Schwannian stroma-rich/stroma-dominant and stroma-poor). This tumor is composed of biologically different clones, an aggressive clone composed of grossly visible, hemorrhagic neuroblastic nodules (stroma-poor component), and a nonaggressive clone comprising ganglioneuroblastoma, intermixed (stroma-rich component), or with ganglioneuroma (stroma-dominant component). The fourth category is ganglioneuroma (Schwannian-stroma–dominant), of which there are two subtypes, maturing and mature. The maturing subtype is composed predominantly of ganglioneuromatous stroma with scattered differentiating neuroblasts or maturing ganglion cells, as well as fully mature ganglion cells. The mature subtype is composed of ganglion cells and Schwannian stroma.
| International Neuroblastoma Pathology Classification | Original Shimada Classification | Prognostic Group | |
|---|---|---|---|
| Neuroblastoma | Schwannian Stroma-poor * | Stroma-poor | |
| Favorable | Favorable | Favorable | |
| <1.5 yr | Poorly differentiated or differentiating and low or intermediate MKI tumor | ||
| 1.5-5 yr | Differentiating and low MKI tumor | ||
| Unfavorable | Unfavorable | Unfavorable | |
| <1.5 yr |
| ||
| 1.5-5 yr |
| ||
| ≥5 yr | All tumors | ||
| Ganglioneuroblastoma -Nodular | Composite Schwannian stroma rich/stroma dominant with stroma poor | Stroma-rich nodular (Unfavorable) | Unfavorable |
| Ganglioneuroblastoma-intermixed | Schwannian stroma rich | Stroma rich intermixed (Favorable) | Favorable |
| Ganglioneuroma | Schwannian stroma dominant | ||
| Maturing | Well differentiated (Favorable) | Favorable ‡ | |
| Mature | Ganglioneuroma | ||
* Subtypes of neuroblastoma are described in detail elsewhere.
† Rare subtype, especially diagnosed in this age group. Further investigation and analysis are required.
‡ Prognostic grouping for these tumor categories is not related to patient age.
In the new and revised International Neuroblastoma Risk Group (INRG) classification schema, tumor histology will be classified independent of age and will primarily be based on degree of differentiation and the MKI.
Clinical Presentation
Neuroblastomas are tumors of sympathetic nervous system and can arise anywhere along the sympathetic chain or in any sympathetic ganglia. Most primary tumors occur in the abdomen (65%), and half of abdominal tumors occur in the adrenal gland. Other common sites of disease origin include the chest, neck, and pelvis, although rarely a primary tumor cannot be found. Because the sites of origin of neuroblastoma are so diverse, the signs and symptoms of disease at presentation vary widely and depend on both the location of the primary tumor and the degree of disease dissemination.
Localized Disease
Approximately 40% of patients present with localized disease. Primary abdominal disease can present as an asymptomatic abdominal mass or with abdominal pain and fullness. Symptoms of obstruction can occasionally be seen, along with renin-mediated hypertension caused by compression of the renal vasculature. Abdominal or pelvic tumors can also occasionally cause compression of lower extremity venous and lymphatic drainage, leading to lower extremity and scrotal swelling. Rarely abdominal tumors spontaneously hemorrhage, and patients with such hemorrhage present with a sudden, dramatic enlargement of an abdominal mass with increased distension and pain. Lower thoracic tumors are usually identified incidentally when a chest radiograph is obtained for unrelated reasons. Occasionally large thoracic tumors are associated with mechanical obstruction and resultant superior vena cava syndrome. Upper thoracic or cervical tumors may cause Horner syndrome. Congenital tumors arising in this site can also cause hematochromia of the iris as a result of decreased pigmentation of the iris on the affected side. Neuroblastoma arising from the paraspinal ganglia in the chest, abdomen, or pelvis can grow through the intervertebral foramina and compress the spinal cord. Patients may be asymptomatic or present with pain or neurologic deficits resulting from spinal cord compression, which requires emergent treatment as detailed later in this chapter.
Metastatic Disease
Metastatic spread of neuroblastoma occurs through the lymphatics or hematogenously. Regional lymph node metastases are seen in one third of patients with apparently localized tumors, and half of patients present with hematogenous metastases. Hematogenous spread occurs most frequently to the bone, bone marrow, and/or liver; rarely, metastasis to the lungs and brain occurs, usually at relapse rather than at presentation. Classic signs of metastatic neuroblastoma include proptosis and periorbital ecchymoses (commonly referred to as “raccoon eyes”) due to tumor infiltration of the periorbital bones. Patients also frequently present with limping and irritability due to bone pain from bone and bone marrow disease, as well as nonspecific symptoms including fever and failure to thrive. The presence of fever is usually associated with extensive bone metastases. Signs and symptoms of bone marrow replacement are also sometimes seen—most frequently pallor (which may also be caused by bleeding within the primary tumor), as well as bruising and increased risk of infection as a result of a low white blood cell count.
Stage 4S Neuroblastoma
Stage 4S (S = special) is a unique presentation of neuroblastoma seen in infants; it was originally described by D’Angio, Evans, and Koop in 1971. Infants with stage 4S neuroblastoma often present with massive hepatomegaly caused by diffuse liver metastases and nontender, bluish subcutaneous nodules as a result of skin nodules. Stage 4S disease accounts for 7% to 12% of neuroblastoma diagnoses and is historically defined as the presence of a small, localized primary tumor with metastases isolated to the liver, skin, and/or bone marrow in an infant younger than 12 months. This special neuroblastoma often spontaneously regresses, although infants younger than 2 months can present with respiratory compromise due to a rapidly enlarging liver and may require cancer-directed treatment. Based on an analysis of a large cohort of international patients diagnosed with neuroblastoma between 1990 and 2002, the INRG Task Force has extended the definition of 4S neuroblastoma to include patients up to the age of 18 months and those with primary tumors crossing the midline.
Paraneoplastic Syndromes
Opsoclonus-myoclonus syndrome (OMS) and vasoactive intestinal peptide syndrome, which are well-described paraneoplastic syndromes associated with neuroblastoma, are detailed in this section, along with a newly described syndrome known as rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation (ROHHAD). Patients with these syndromes usually have localized tumors with favorable biology, perhaps because the syndrome causes early detection of tumor or, more likely, because of a correlation between the syndrome and the tumor biology.
OMS is observed in 2% to 3% of children with neuroblastoma and is usually diagnosed between the age of 1 and 3 years. It is often referred to as “dancing eyes, dancing feet syndrome” and is characterized by the acute onset of rapid eye movements, ataxia, myoclonic jerking of the limbs and trunk, and behavioral disturbances. Although children with neuroblastoma and OMS generally have favorable tumor prognostic features and a high survival rate, 70% to 80% experience long-term neurologic sequelae that can include global developmental delay, speech and/or motor delay, behavioral disturbances, and cognitive deficits that can seriously affect quality of life. OMS in children is associated with neuroblastoma more than half of the time. After exclusion of CNS pathology, all children with OMS should be evaluated for neuroblastoma with imaging of their chest and abdomen, an iodine 123–metaiodobenzylguanadine (MIBG) scan, and urine catecholamines. Traditional methods to detect neuroblastoma may be insensitive in patients with OMS, and therefore high-resolution computed tomography (CT) or magnetic resonance imaging (MRI) of the chest and abdomen should be considered, even in patients with negative catecholamines or negative results of an MIBG scan, to screen for small tumors.
OMS and its sequelae are thought to be immune mediated and are likely caused by the presence of an antineural antibody that cross-reacts with a common antigen on both neuroblastoma and normal nervous system tissue. Improvement in symptoms can be seen after tumor removal in some cases, but generally immunosuppression with glucocorticoids or adrenocorticotropic hormone is used to relieve acute symptoms. However more than 80% of patients will have a relapse of symptoms with steroid weaning or in association with a viral syndrome, and more than half of patients will need prolonged steroid treatment, necessitating additional treatment approaches. Intravenous immune globulin has also been used in the treatment of OMS, either with or without steroids, with varying success. Case reports have shown efficacy of other immunomodulating strategies including plasmapheresis, rituximab, and mycophenolate. Although often effective in treating acute symptoms, none of these treatments has been consistently correlated with improved long-term outcome. Interestingly, a report from the Pediatric Oncology Group (POG) suggested that patients who received chemotherapy to treat their neuroblastoma had a more favorable neurologic outcome. This benefit may be due to the immunosuppressive effects of chemotherapy or to less severe activation of the immune system in patients with more advanced stages of tumor. The same benefit was not observed in a similar review performed by the Children’s Cancer Group (CCG), and the benefit of chemotherapy for the treatment of OMS needs to be investigated further. The COG recently conducted a trial for OMS in which patients with low-risk neuroblastoma who would not otherwise receive chemotherapy were treated with cyclophosphamide in addition to steroids. Long-term outcome of these patients compared with historic control subjects should provide insight regarding the role of chemotherapy for the treatment of OMS.
Further investigation is needed to determine the best treatment approach for OMS. Regardless of treatment, physicians caring for patients with OMS should anticipate long-term neurologic abnormalities and use early intervention to help minimize these deficits.
Tumor secretion of vasoactive intestinal peptide can cause a syndrome of chronic watery diarrhea and failure to thrive. Most tumors secreting vasoactive intestinal peptide are histologically mature. Surgical removal of the tumor usually results in complete resolution of symptoms.
ROHHAD syndrome is characterized by rapid-onset obesity, hypothalamic dysfunction, hypoventilation, and autonomic dysregulation. Tumors of neural crest origin have been found in more than one third of cases and are usually differentiated (ganglioneuromas or ganglioneuroblastomas). ROHHAD syndrome most commonly affects otherwise normal children between 2 and 4 years of age and typically presents first with rapid weight gain and obesity followed at varying time intervals by autonomic dysfunction and hypoventilation. The prognosis is poor, and at least 25% of children die because of respiratory failure. A genetic cause has been suggested because of the constellation and consistency of symptoms and the reported occurrence in siblings, but several candidate genes (including PHOX2B, ASCL1 [achaete-scute family bHLH transcription factor–1] , NTRK2 [TRKB] , and BDNF ) have been sequenced in affected patients and have not been informative. Monozygotic twins discordant for ROHHAD have recently been reported, although this in itself should not preclude a genetic cause because modifier genes or epigenetic variation may play a role in the variable phenotype. Because of the association with neuroblastic tumors and the finding of extensive infiltrates of lymphocytes and histiocytes in the hypothalamus of some patients, a paraneoplastic cause has been postulated. Immune-modulating agents including intravenous immunoglobulin, rituximab, and cyclophosphamide have been tried in small series of patients with varying success, and collaborative studies are needed to determine the cause of this rare disorder. Surveillance for tumors in patients with this constellation of features is recommended.
Diagnosis and Evaluation
Diagnosis
The diagnosis of neuroblastoma is usually established from histopathologic evaluation of primary tumor tissue. In most cases, especially if features of neuronal differentiation are present, a tissue diagnosis of neuroblastoma can be made on the basis of conventional hematoxylin and eosin staining. However, in cases in which there is little differentiation and only small round blue cells are evident, immunohistochemical staining for neuron-specific enolase, chromogranin A, and/or synaptophysin, as well as cytogenetic and molecular analysis, can help differentiate neuroblastoma from other small round blue cell tumors of childhood.
In addition to establishing the diagnosis, primary tumor material is essential for risk stratification and prognosis, particularly in children younger than 18 months with locoregional or metastatic spread. In cases in which primary tumor tissue cannot be obtained safely, the diagnosis of neuroblastoma can also be made by demonstrating unequivocal neuroblastoma cells in a bone marrow aspirate or biopsy in conjunction with increased urinary catecholamines. Urinary catecholamine metabolites are increased in 90% to 95% of neuroblastomas using sensitive detection techniques such as high-performance liquid chromatography and can be helpful in establishing a diagnosis, as well as in monitoring disease activity and response to therapy in patients who are known to excrete the metabolites. In sympathetic cells the catecholamine precursor 3,4-dihydroxyphenylalanine (DOPA) is converted to dopamine by DOPA decarboxylase, which is converted to norepinephrine and then to epinephrine by the enzyme phenylethanolamine-N-methyltransferase, which is not present in neuroblastoma cells. Instead the enzymes catechol-O-methyltransferase and monoamine oxidase convert DOPA and dopamine to homovanillic acid and norepinephrine and epinephrine to vanillylmandelic acid, both of which are inactive metabolites that are excreted in the urine. Both urinary homovanillic acid and vanillylmandelic acid should be measured for diagnostic purposes. For undifferentiated tumors, urinary dopamine may also be useful. Despite this catecholamine production, symptoms of catecholamine excess such as hypertension, flushing, and sweating are rarely seen in persons with neuroblastoma, likely because of the relatively low concentrations of active catecholamines in the circulation.
Clinical Disease Assessment
The primary tumor and the extent of disease should be evaluated using imaging techniques both at diagnosis and to evaluate response to treatment. Either CT or MRI are routinely used to evaluate the extent and origin of the primary tumor, as well as possible solid organ metastases. CT is more widely available and can rapidly capture images, reducing the need for sedation, but it is associated with substantial radiation exposure. Ionizing radiation is not used to perform MRI but it has a longer imaging acquisition time, making sedation necessary in most young children. Currently no consensus exists with regard to which imaging technique is optimal for evaluating tumors and metastases in the abdomen, pelvis, or mediastinum. Availability and clinical factors, including the risks of sedation, should be taken into account. MRI is preferred for assessing paraspinal lesions, particularly those with possible intraextension and spinal cord impingement. Although abdominal ultrasonography is not as useful for assessing tumor volume and anatomy, it may be useful as a noninvasive method for following tumor response or surveying for disease relapse. Brain imaging is only recommended if clinically indicated by symptoms or examination.
Bone and bone marrow should also be evaluated by imaging to look for the presence and extent of metastatic disease. MIBG is a norepinephrine analogue that is selectively concentrated in more than 90% to 95% of neuroblastomas and can be combined with either the iodine 131 or I isotopes for scanning purposes. MIBG scintigraphy is highly sensitive and specific for detecting osteomedullary disease and can also be used to assess the primary tumor, as well as occult soft tissue disease ( Fig. 54-5 ). The I isotope provides enhanced image resolution and is the isotope of choice when it is available. MIBG scans are essential for the initial staging of neuroblastoma and should be performed prior to primary tumor resection. International guidelines are available for patient preparation, radiotracer administration, and imaging techniques. A bone scan using technetium 99m–diphosphonate scintigraphy is a relatively sensitive but less specific method to detect bony metastases in patients with neuroblastoma. Bone scans are recommended in cases in which the primary tumor is not MIBG avid, or in cases in which tumor uptake of MIBG cannot be confirmed (i.e., in cases in which the primary tumor is removed prior to imaging for metastatic disease). Fluorine 18–labeled deoxyglucose positron emission tomography (FDG-PET) can also be used to evaluate metastatic disease in persons with neuroblastoma. Its sensitivity and specificity in comparison with MIBG scintigraphy is still being evaluated, with studies suggesting that MIBG is more sensitive than FDG-PET for the detection of bone lesions and skull-based lesions, and that FDG-PET is more sensitive than MIBG for the detection of small soft tissue tumors and nodal metastases. The routine use of FDG-PET is not currently recommended, but it may be a helpful tool to follow-up patients with metastatic disease who do not have MIBG-avid tumors.
Bone marrow aspirates and biopsies from both iliac crests are recommended to assess for the presence of bone marrow disease using standard histologic analysis. Both immunocytochemical and polymerase chain reaction–based technologies can be used to increase sensitivity of marrow detection. Immunocytochemical analysis of bone marrow aspirates with monoclonal antibodies directed against neural-specific antigens (e.g., GD2 and NCAM, neural cell adhesion molecule) increases the sensitivity of detecting marrow involvement to 1 : 100,000 nucleated cells. Reverse-transcriptase polymerase chain reaction methodologies that target the expression of neuroblastoma-specific messages such as tyrosine hydroxylase, protein gene product 9.5, or GD2 synthase can enhance sensitivity further. However, the clinical and prognostic significance of this enhanced detection, remains to be determined, and these studies are generally only recommended within the context of a clinical study.
Staging
The International Neuroblastoma Staging System (INSS) was established in the 1990s to replace three major staging systems used throughout the world in an effort to provide international uniformity so that clinical trials and biology studies performed by different groups in different countries could be compared. The INSS ( Table 54-3 ) is largely based on the surgical assessments of a patient at diagnosis and has been used nationally and internationally for more than 20 years. However, because surgical approaches are not uniform, the INSS stage for patients with locoregional disease can vary considerably depending on the degree of initial tumor resection, making comparison of clinical trials conducted in different parts of the world difficult. To address this problem, the INRG Task Force developed a new staging system based on tumor imaging rather than extent of surgical resection. This staging system (known as the INRG staging system) uses radiologic characteristics of the primary tumor to predict surgical risk and resectability ( Tables 54-4 and 54-5 ). Using this approach, in INRG staging, locoregional disease is classified as stage L1 or L2 on the basis of whether the tumor is locally invasive by imaging at the time of diagnosis. Stage M indicates metastatic disease that is assessed in the same way it was assessed by INSS criteria, via both imaging and pathology.
| Stage | Description |
|---|---|
| 1 | Localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes negative for tumor microscopically (nodes attached to and removed with the primary tumor may be positive) |
| 2A | Localized tumor with incomplete gross excision; representative ipsilateral nonadherent lymph nodes negative for tumor microscopically |
| 2B | Localized tumor with or without complete gross excision, with ipsilateral nonadherent lymph nodes positive for tumor; enlarged contralateral lymph nodes must be negative microscopically |
| 3 | Unresectable unilateral tumor infiltrating across the midline, * with or without regional lymph node involvement; or localized unilateral tumor with contralateral regional lymph node involvement; or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement |
| 4 | Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin and/or other organs (except as defined for stage 4S) |
| 4S | Localized primary tumor (as defined for stage 1, 2A, or 2B), with dissemination limited to skin, liver and/or bone marrow † (limited to infants younger than 1 year of age) |
* The midline is defined as the vertebral column. Tumors originating on one side and “crossing the midline” must infiltrate to or beyond the opposite side of the vertebral column.
† Marrow involvement in stage 4S should be minimal, that is, less than 10% of total nucleated cells identified as malignant on bone marrow biopsy or aspirate. More extensive marrow involvement would be considered to be stage 4. The metaiodobenzylguanidine scan (if done) should be negative in the marrow.
| Anatomic Region | Description |
|---|---|
| Multiple body compartments | Ipsilateral tumor extension within two body compartments |
| Neck | Tumor encasing carotid artery, vertebral artery, and/or jugular vein |
| Tumor extending to skull base | |
| Tumor compressing trachea | |
| Cervicothoracic junction | Tumor encasing brachial plexus roots |
| Tumor encasing subclavian vessels, vertebral artery, and/or carotid artery | |
| Tumor compressing trachea | |
| Thorax | Tumor encasing aorta and/or major branches |
| Tumor compressing trachea and/or principal bronchi | |
| Lower mediastinal tumor infiltrating costovertebral junction between T9 and T12 vertebral levels | |
| Thoracoabdominal junction | Tumor encasing aorta and/or vena cava |
| Abdomen and pelvis | Tumor infiltrating porta hepatis and/or hepatoduodenal ligament |
| Tumor encasing branches of the superior mesenteric artery at the mesenteric root | |
| Tumor encasing the origin of the celiac axis and/or the origin of the superior mesenteric artery | |
| Tumor invading one or both renal pedicles | |
| Tumor encasing the aorta and/or vena cava | |
| Tumor encasing the iliac vessels | |
| Pelvic tumor crossing the sciatic notch | |
| Intraspinal tumor extension | Intraspinal tumor extension provided that more than one third of the spinal canal in the axial plane is invaded, the perimedullary leptomeningeal spaces are not visible, or the spinal cord signal intensity is abnormal |
| Infiltration of adjacent organs and structures | Pericardium, diaphragm, kidney, liver, duodenopancreatic block, and mesentery |
| Stage | Description |
|---|---|
| L1 | Localized tumor not involving vital structures as defined by the list of image-defined risk factors and confined to one body compartment |
| L2 | Locoregional tumor with the presence of one or more image-defined risk factors |
| M | Distant metastatic disease (except stage MS) |
| MS | Metastatic disease in children younger than 18 months with metastases confined to skin, liver, and/or bone marrow |
The INRG system also differs from the INSS system in that the midline and lymph node status are not included in the staging criteria. In addition, INRG stage MS includes patients up to 18 months of age and those with L2 tumors, whereas INSS stage 4S has an upper age limit of 12 months and includes only patients with stage 1 or 2 primary tumors.
Because imaging can be retrospectively reviewed, the INRG staging system should provide data that are more uniform than the INSS system. The INRG staging system is currently used in Europe and will be adopted into the COG studies as well.
Risk Stratification
Neuroblastoma is a tumor in which biologic factors are consistently used to influence disease risk stratification and treatment. In 1998 the COG established a risk-stratification system that stratifies patients into low-, intermediate-, or high-risk categories based on prognostic features including age at diagnosis, stage, tumor histology, DNA index (ploidy), and MYCN amplification status ( Table 54-6 ). Treatment recommendations are based on this risk grouping, as described in the next section, and prognosis is distinct for each risk group ( Fig. 54-6 ).
| INSS | Age | MYCN * | Ploidy | Shimada † | Risk Group |
|---|---|---|---|---|---|
| 1 | Any | Any | Any | Any | Low |
| 2 | Any | Not amp | Any | Any | Low |
| 2 | Any | Amp | Any | Any | High |
| 3 | <547 days | Not amp | Any | Any | Intermediate |
| 3 | ≥547 days | Not amp | Any | FH | Intermediate |
| 3 | ≥547 days | Not amp | Any | UH | High |
| 3 | Any | Amp | Any | Any | High |
| 4 | <365 days | Amp | Any | Any | High |
| 4 | <365 days | Not amp | Any | Any | Intermediate |
| 4 | 365 – <547 days | Not amp | >1 | FH | Intermediate |
| 4 | 365 – <547 days | Any | 1 | Any | High |
| 4 | 365 – <547 days | Any | Any | UH | High |
| 4 | 365 – <547 days | Amp | Any | Any | High |
| 4 | ≥547 days | Any | Any | Any | High |
| 4S | <365 days | Not amp | 1 | FH | Low |
| 4S | <365 days | Not amp | 1 | Any | Intermediate |
| 4S | <365 days | Not amp | Any | UH | Intermediate |
| 4S | <365 days | Amp | Any | Any | High |
* MYCN amplification status: Amp, amplified; non-amp, not amplified.
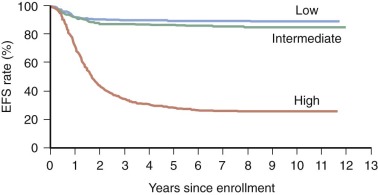
In an effort to unify risk stratification internationally and facilitate comparison of clinical trials performed in different parts of the world, the INRG Task Force was formed with representatives from the major pediatric cooperative groups. The INRG Task Force retrospectively analyzed the prognostic effect of 13 variables in a cohort of 8800 patients diagnosed with neuroblastoma between 1990 and 2002 with respect to EFS. Based on these analyses, a consensus INRG classification scheme was created using the most statistically significant and clinically relevant factors, which include INRG stage, age, histology, grade of tumor differentiation, MYCN and 11q status, and tumor ploidy ( Table 54-7 ). The significance of the risk factors used in both the INRG and COG risk stratification systems is described later.
| INRG Stage | Age (mo) | Histologic Category | Grade of Tumor Differentiation | MYCN | 11q Aberration | Ploidy | Pretreatment Risk Group |
|---|---|---|---|---|---|---|---|
| L1/L2 | GN maturing; GNB intermixed | A Very low | |||||
| L1 | Any, except GN maturing or GNB intermixed | NA | B Very low | ||||
| Amp | K High | ||||||
| L2 | <18 | Any, except GN maturing or GNB intermixed | NA | No | D Low | ||
| Yes | G Intermediate | ||||||
| ≥18 | GNB nodular; neuroblastoma | Differentiating | NA | No | E Low | ||
| Yes | H Intermediate | ||||||
| Poorly differentiated or undifferentiated | NA | ||||||
| Amp | N High | ||||||
| M | <18 | NA | Hyperdiploid | F Low | |||
| <12 | NA | Diploid | I Intermediate | ||||
| 12 to <18 | NA | Diploid | J Intermediate | ||||
| <18 | Amp | O High | |||||
| ≥18 | P High | ||||||
| MS | <18 | NA | No | C Very low | |||
| Yes | Q High | ||||||
| Amp | R High |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


