Skeletal Muscle, Aging, and Disease
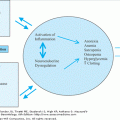
Skeletal muscle is a large and important organ system in the human body. Disorders and diseases of muscle are common in late life and have a major impact on function and quality of life. Many diseases affect muscle and are considered forms of myopathy. Myopathies are generally considered to be inflammatory or noninflammatory and will be addressed below. Loss of skeletal muscle mass owing to any disease or condition is called sarcopenia. Sarcopenia derives from the Greek and translates as “poverty of flesh.” As a medical term, sarcopenia is nonspecific and can be caused by aging, disuse, wasting illness, or starvation or can be a secondary consequence of ischemia or neuropathy. Table 119-1 describes the characteristics of each of these forms of sarcopenia and highlights implications for care of the older adult. Sarcopenia can also develop in the course of some myopathies. Sarcopenia caused by aging itself is difficult to characterize since aging is often accompanied by the other contributors to loss of muscle described in Table 119-1.
INTRINSIC MUSCLE AGING | DECONDITIONING/DISUSE | WASTING OWING TO CANCER, HIV, INFLAMMATORY CONDITIONS, OR CHRONIC DISEASES | STARVATION (PROTEIN MALNUTRITION) OR INCREASED METABOLIC DEMANDS | ISOLATED MUSCLE LOSS OWING TO LOCALIZED NERVOUS SYSTEM DISEASE OR CARDIOVASCULAR DISEASE | |
|---|---|---|---|---|---|
Myopathy | Not a myopathy, considered normal | Yes, considered abnormal | Yes, considered abnormal | Yes, considered abnormal | Yes, considered abnormal |
Etiology | Normal intrinsic aging process | Loss of stimulation to muscle protein synthesis and maintenance of neuromuscular junctions. Similar to effects of low-gravity environment. | Increased inflammatory markers | Lack of sufficient protein reserves to counteract for muscle catabolism. Fasting results in decreased synthesis and increased breakdown of both myofibrillar and soluble muscle proteins and creates alternative sources of energy. | Peripheral vascular disease with consequent ischemia of an extremity, severance of a specific peripheral nerve with paralysis, cardiovascular accidents with paresias or paralysis, and loss of neuromuscular junctions because of similar conditions. |
Creatinine kinase (CK) | Normal | Normal | Normal | Normal | Normal, unless there is an infarct of the region. |
Potential implications for aging | Isolated effects of aging are hard to define, since most older adults also have other conditions that contribute to sarcopenia. The muscular system is integrated with other organ systems and is affected by disorders in other systems. | Levels of activity tend to decrease with aging. Many diseases reduce activity and cause secondary deconditioning. | Many wasting diseases increase in prevalence with age. | Calorie and protein intake can decrease with age and many conditions. Gastrointestinal absorption of nutrients may also decrease owing to several conditions that occur in older adults. | Cardiovascular and neurological conditions are common in older adults. |
Usual presentation | Very chronic across decades of later life | Acute, subacute, or chronic | Acute, subacute, or chronic | Rarely acute. Usually subacute and chronic | Acute or subacute onset, eventually chronic |
Caloric ingestion | Normal | Normal | Normal | Abnormal | Normal |
Usual age of presentation | Starts around fourth decade of life | Any age | Any age | Any age | Any age |
Approximate rate of loss | ~1% per year | ~1% per day | ~1% per day | ~1% per day | ~1% per day |
Usual outcome | Irreversible | Reversible | Potentially reversible | Reversible | Irreversible, partially reversible, or potentially reversible in rare cases |
Histology | Muscle atrophy with no evidence of necrosis: loss of muscle cells, decrease in the size of existing muscle cells. Affecting type 2B cells more at the beginning, then affecting all types. | Muscle atrophy with no evidence of necrosis: decrease in size of muscle cells (fiber). | Muscle atrophy with no evidence of necrosis: decrease in the size of existing muscle cells. There could be loss of muscle cells. | Muscle atrophy with no evidence of necrosis: decrease in the size of existing muscle cells. | Muscle atrophy unless there is an infarction where muscle necrosis ensues. |
Treatment | No cure. However, exercise can hypertrophy existing muscle cells and improve muscle function to compensate for the decrease in muscle cells and minimize the clinical impact of the muscle loss over time. | Aerobic and anaerobic exercise. Encourage physical therapy and occupational therapy. | Correction of the underlying cause and aerobic and anaerobic exercises | Nutritional correction and aerobic and anaerobic exercises | Rehabilitation exercise for recovery of remaining upper motor neurons for plasticity as in the case of stroke. Stent placement or angioplasty or surgery in the case of peripheral vascular disease. Aerobic and anaerobic exercise. |
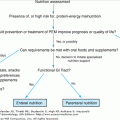
Sarcopenia is now definable based on formal assessment of body composition using dual-energy x-ray absorptiometry. Similar to the use of relative bone mass to make the diagnosis of osteoporosis, dual-energy x-ray absorptiometry can yield estimates of lean body mass, which can be reported as absolute values or as scores relative to normal gender-specific values and skeletal size. In order to adjust for differences in skeletal size, the “relative skeletal muscle index” (RSMI) is defined as the predicted or measured (dual-energy x-ray absorptiometry) muscle mass (kg) divided by stature in square meters (RSMI = kg/m2). On this scale, sarcopenia is defined by index values less than 2 standard deviations below the gender-specific mean for RSMI in a healthy, younger population. Thus, cutoff values for sarcopenia would be less than 7.26 kg/m2 for males and 5.45 kg/m2 for females. Since aging tends to alter body composition, with reduced muscle and bone and increased extracellular fluid volumes, the prevalence of sarcopenia increases with age. Sarcopenia defined as RSMI less than 2 standard deviations below normal occurs in approximately 15% of people aged 60 to 69 years and approximately 40% of people older than 80 years.
Myopathies
Several of the most important diseases that primarily affect muscle are inflammatory in nature, are together termed idiopathic inflammatory myopathies, and carry labels using the term “myositis.” The most common forms include polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM). Each will be described in the sections below. Since all three are often treated with glucocorticoids, a separate section discusses steroid treatment.
PM is an IIM that affects all ages, with approximately one-third of cases in persons older than 65 years. Females are affected more than males. While the exact pathophysiology is not known, muscle biopsy shows inflammation that is mediated by T cells, mostly CD8+ T cells and macrophages causing perimysial and perivascular infiltrations. The inflammatory cells surround and invade non-necrotic muscle fibers, leading to necrosis at various stages alternating with regeneration and replacement by fibrous connective tissue and fat.
PM usually presents in an insidious fashion, and the course may be subacute or chronic with either no or mild pain. Common systemic features include fatigue, weight loss, arthralgias, and fever. Skin sensation and reflexes are typically normal. Symmetrical proximal weakness manifests in the neck flexors and extensors, as well as the shoulders and hips; dysphagia may occur in 25% of cases. Creatinine kinase (CK) is elevated in 90% of cases, but myoglobinuria is usually not present. Anti-Jo-1 or anti-Mi2 antibodies may be present. Patients usually present with anemia and positive inflammatory markers. PM may be associated with inflammation in other organ systems such as pulmonary interstitial fibrosis or myocarditis and is sometimes a component of other connective tissue disorders. The electromyogram is abnormal in 90% of cases and shows motor unit action potentials (MUAPs) that are of short duration, of small amplitude, or polyphasic with early recruitment. Fibrillations and positive waves indicate active inflammation.
PM is treated mainly with steroids and cytotoxic agents. More detail about treatment is presented in the section below. With or without treatment, the disease may progress to dysphagia and respiratory failure. Prognosis for recovery is poorer in older adults, in part because of the effect of age on susceptibility to the complications of immunosuppressive drugs.
DM is an IIM that affects all ages, with approximately one-quarter of cases developing in older adults. It has a female preponderance. While the pathophysiology is not fully understood, muscle biopsy shows endomysial inflammation with a more humoral component (CD4+ T cells and B cells). Perifascicular atrophy (atrophic fibers at the edges of the fascicles) may be caused by hypoperfusion, since capillary density is significantly reduced. Vessels are positive for complement membrane attack complex.
Presentation is insidious and the course is subacute with systemic features such as fatigue, weight loss, arthralgias, and fever. Pain is absent or mild. Sensation and reflexes are preserved. Photosensitivity rashes are common. Typical skin changes include heliotrope rash, Gottron’s papules, V sign, Shawl’s sign, and mechanic’s hands. Heliotrope rash is a violet or purplish discoloration seen around the eyes and on the extremities. Gottron’s papules are erythematous, raised areas at the bony prominences in the hands, elbows, and knees. V sign is a macular erythema on the chest in a V shape. Shawl sign is a macular erythema over the posterior thorax. Mechanic’s hands are rough, cracked regions of skin on the palms and radial aspects of the fingers. Dysphagia occurs in approximately 25% of DM cases. Symmetrical proximal weakness appears in the neck flexors and extensors, as well as shoulders and hips. CK is elevated in 90% of cases, and myoglobinuria is usually not present. Jo-1 antibodies are associated with pulmonary interstitial fibrosis, and anti-Mi2 antibodies are associated with classic DM. Patients usually present with anemia and positive inflammatory markers. DM is sometimes associated with myocarditis and other connective tissue disorders. Electromyograms for DM and PM are indistinguishable.
Treatment involves steroids and cytotoxic agents. The disease sometimes progresses to dysphagia and respiratory failure. Prognosis, even with treatment, is worse for older than younger adults. Routine cancer screening is required, since 25% of cases with DM develop some type of associated cancer (mostly colon).
IBM is an IIM with a male preponderance and is the most common IIM in older adults. While the pathophysiology is not known, muscle biopsy shows endomysial infiltrates with CD8+ T cells, macrophages invading non-necrotic muscle fibers, as well as rimmed vacuoles and amyloid deposits in the muscle fibers.
The onset of IBM is more insidious than PM and DM, and its course is either subacute or chronic. Pain is absent or mild. Reflexes are either diminished or absent as a result of associated peripheral neuropathy. As opposed to the proximal muscle weakness that predominates in PM and DM, in IBM the proximal and distal muscles are equally involved. IBM might typically present with weakness of bilateral wrists, finger flexors, quadriceps, and foot dorsiflexors. IBM can present with atrophy of the quadriceps muscles. CK elevation in IBM is less than that for PM and DM. Skin involvement and myoglobinuria are usually not present.
The electromyogram shows a chronic myopathy with an active component (fibrillation). Fibrillations and positive waves indicate active inflammation. Nerve conduction studies reveal mild neurogenic features.
IBM is typically unresponsive to steroids. Up to one-third of cases will stabilize or improve with or without treatment. IBM medications, when used, are similar to those for PM and DM.
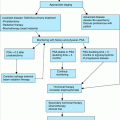
Glucocorticoids are the first-line treatment for inflammatory myopathies. Initial dosage can be reduced for older compared to young adults, because overall muscle mass is expected to be lower with age. While prednisone dosing in young adults is calculated at 1.5 mg/kg (up to 100 mg/day), the dose in older adults is calculated at 1.0 mg/kg (up to 60 mg/day). The initial dose should be maintained until there is evidence of clinical response, especially a clinically detectable improvement in muscle strength. If there is no clinical response in 3 or 4 months, the patient should be considered steroid unresponsive and another immunosuppressive agent should be considered. Tapering begins with clinical response. Dosing can switch immediately to an alternate-day pattern (e.g., 60 mg every other day) or can be gradually converted to an alternate-day pattern (e.g., reduce the alternate or “off-day” dose by 10 mg per week). Continue the alternate-day regimen until either the patient’s strength is normalized or improvement reaches a plateau (usually 4–6 months). Next, reduce the dosage by 5 mg every 2 to 3 weeks until the lowest dose capable of controlling symptoms and maintaining muscle strength is achieved. A maintenance dose of 10 to 25 mg every other day may be needed to achieve stability. This approach to tapering steroids can also apply to other inflammatory conditions such as polymyalgia rheumatica (PMR) and temporal arteritis.
Glucocorticoids are the only agents approved by the U.S. Food and Drug Administration (FDA) for treatment of inflammatory myopathies. The efficacy of other immunosuppressive agents in inflammatory myositis is unproven, and a recent systematic review found insufficient high-quality randomized controlled trials to make evidence-based conclusions. However, second-line use of other immunosuppressive agents might be considered in several circumstances. First, second-line agents might be helpful when a steroid-sparing effect is needed to avoid complications associated with steroid therapy. Second, such agents might be used when a relapse occurs repeatedly after attempts to taper steroids. Third, second-line agents might be attempted when first-line therapy with steroid has failed to show a clinical response after at least 3 months. Fourth, second-line agents might be added to steroids when the disease is progressing rapidly, with life-threatening manifestations such as severe weakness and/or respiratory failure. Table 119-2 summarizes the available agents. Some of these agents are very expensive, and cost versus likelihood of benefit and harm should be carefully considered. It is good practice to involve the patient, and the family if the patient desires, in discussions about treatment options.
Drug | Usual Dose | Main Side Effects |
|---|---|---|
Methotrexate | 7.5–20 mg oral or injection dose weekly | Diarrhea, mucosal ulcers, bone marrow toxicity, liver toxicity, and interstitial pneumonitis. Avoid methotrexate if patient has Jo-1 antibodies. Increased risk of opportunistic infections. |
Azathioprine | 50–150 mg oral dose daily | Bone marrow toxicity and liver toxicity. Increased risk of opportunistic infections. |
Mycophenolate mofetil | 2–3 g oral dose daily | Diarrhea and bone marrow toxicity. Increased risk of opportunistic infections. |
Cyclophosphamide | 50–150 mg oral dose daily | Nausea, vomiting, alopecia, bone marrow toxicity, and hemorrhagic cystitis. Increased risk of opportunistic infections. |
Intravenous immunoglobulin (IVIg) |