Myeloproliferative disorders (MPD) are chronic diseases caused by clonal proliferation of bone marrow stem cells usually leading to excess production of one or more haemopoietic lineage. The clinical syndromes include polycythaemia rubra vera (PRV) (red cells), essential thrombocythaemia (ET) (platelets) and primary myelofibrosis in which there is a reactive fibrosis of the marrow and extramedullary haemopoiesis in the liver and spleen. Intermediate forms may occur and the diseases may all transform into acute myeloid leukaemia. A mutation (Val617Phe) of the Janus kinase 2 (JAK2) is present in nearly 100% of cases of PRV and about 50% of cases of ET or primary myelofibrosis (Fig. 26.1). It is usually heterozygous, but homozygous in 5–10% of positive cases, and these tend to be clinically more severe. The mutation causes autonomous activation of signal transduction from the growth factor receptor for erythropoietin. This leads to uncontrolled cell proliferation. Chronic myeloid leukaemia was formerly classified as an MPD but is now known to be due to a different mutation (BCR-ABL1; see Chapter 24).
Differential diagnosis
Increased levels of red cells, white cells and platelets can occur in a range of physiolological and reactive conditions. Reactive causes of increased levels of white cells are considered in Chapter 18; secondary or reactive causes of increased levels of red cells, platelets and of increased bone marrow fibrosis are listed in Boxes 26.1–26.3 and shown in Fig 26.2. Careful clinical assessment is required to distinguish these reactive (secondary) conditions from primary MPD in which there is an intrinsic disorder within the marrow stem cells. The general underlying mechanism for the secondary or reactive disorders is that an external stimulus leads to increased elaboration of erythropoietin and/or thrombopoietin (see Chapter 1
Related posts:
 Clinical assessment
Megaloblastic anaemia II: clinical features, treatment and other macrocytic anaemias
Haemolytic anaemias V: genetic defects of haemoglobin – sickle cell disease
Lymphoma II: Hodgkin lymphoma
Stem cell transplantation
Clinical assessment
Megaloblastic anaemia II: clinical features, treatment and other macrocytic anaemias
Haemolytic anaemias V: genetic defects of haemoglobin – sickle cell disease
Lymphoma II: Hodgkin lymphoma
Stem cell transplantation
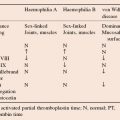 Disorders of haemostasis II: inherited disorders of coagulation
Disorders of haemostasis II: inherited disorders of coagulation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


