Fig. 4.1
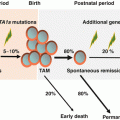
Distribution of patients with myelodysplastic syndrome, myeloproliferative neoplasms, and bone marrow failure in Japan (Reference [3]). One thousand patients were prospectively underwent the central review of morphology and histology from February 2009 to October 2013. This data was provided by the courtesy of Dr. Asahito Hama
Irrespective of subtype, MDS is diagnosed in all age groups with median age at diagnosis ranging from 6 to 12 years from previous studies [9–14]. Boys and girls are equally affected.
4.2.2 Classification
Historically, The French–American–British (FAB) cooperative group produced the first classification of MDS in 1982, based on experiences from adult patients, which divided MDS into five subgroups: refractory anemia (RA), RARS, RAEB, RAEB-T, and CMML [15]. Distinction between these subtypes was largely based on the proportion of blasts in the peripheral blood (PB) and bone marrow (BM) and the degree of monocytosis in the PB. The FAB classification became an important tool of communication about MDS and had a prognostic impact. In 2001, the WHO classification of hematological malignancies incorporates both morphology and cytogenetic changes [16]. The 2001 WHO classification lowered the threshold for distinguishing AML from MDS, from 30 to 20% blasts in the BM. Although JMML was recognized as a separate entity in the WHO classification, both the FAB and the WHO classifications were based on the review of adult cases and did not acknowledge the differences between MDS in children and adults. For example, RARS and the unique 5q-syndrome were extremely rare in children. The significance of multilineage dysplasia and a blast threshold of 20% for distinction between MDS and AML were unknown in children.
Considering these issues, a pediatric approach to the diagnosis and management of MDS and MPD was proposed in 2003 [1]. It divided MDS/MPD in children into three main groups; JMML, myeloid proliferations related to Down syndrome, and MDS (Table 4.1). MDS was further subdivided into refractory cytopenia (RC), RAEB, and RAEB-T. This classification of pediatric MPD/MDS was incorporated into the fourth edition of the WHO classification in 2008 [2]. RCC is defined as a provisional entity characterized by persistent cytopenia with <5% blasts in the BM, <2% blasts in the PB, and dysplastic changes in two or three lineages or exceeding 10% in one single lineage [2]. RAEB is applied to children with MDS in whom there are 2–19% blasts in the PB or 5–19% blasts in the BM, similarly to adult MDS [2]. However, the prognostic significance of subdivision of RAEB into RAEB-1 and RAEB-2 has not been shown in children so far [17]. Children with 20–29% blasts in the PB and/or BM and myelodysplasia often have slowly progressive disease and behave more like MDS than AML; therefore, the subtype of RAEB-T is retained in the 2008 WHO classification of childhood MDS [2]. Children with recurrent cytogenetic abnormalities such as t(8;21)(q22;q22), inv(16)(p13.1q22) or t(16:16)(p13.1;q22), or t(15;17)(q22;q12) should be considered as AML regardless of blast percentage.
Myelodysplastic/Myeloproliferative disease | ||
Juvenile myelomonocytic leukemia (JMML) | ||
Myeloid proliferations related to Down syndrome (DS) | ||
Transient abnormal myelopoiesis (TAM) | ||
Myeloid leukemia of DS (ML-DS) | ||
Myelodysplastic syndrome (MDS) | (PB blasts) | (BM blasts) |
Refractory cytopenia (RC) | <2% | <5% |
Refractory anemia with excess blasts (RAEB)* | 2–19% | 5–19% |
RAEB in transformation (RAEB-T) | 20–29% | 20–29% |
MDS that arises in a previously healthy child are referred to as “primary MDS” and that developing in a child with a known predisposing condition are referred to as “secondary MDS”. Secondary MDS is seen in patients who previously received chemo- or radiation therapy (therapy-related MDS) [18], in patients with IBMFS [19], following acquired aplastic anemia [20], and as familial MDS [21].
MDS associated with Down syndrome has been reported to account for 20–25% of cases of childhood MDS in the past; however, myeloid leukemia in Down syndrome with blasts less than 20% is now thought as distinct entity from other subtypes of childhood MDS and classified as “myeloid leukemia of Down syndrome” regardless of blast percentage [2].
4.2.3 Clinical and Laboratory Characteristics
Children with MDS usually present with symptoms related to cytopenias such as bleeding tendency, infection, and pallor. Some children have no clinical symptoms and can be diagnosed incidentally during a routine work-up. Cytopenia is generally milder in MDS than aplastic anemia (AA). The length of persistent cytopenia should be at least 1 month. Leukocytosis and organomegaly are generally not features of MDS. Table 4.2 shows minimal diagnostic criteria for pediatric MDS [1, 2].
At least two of the following: |
1. Sustained unexplained cytopenia (neutropenia, thrombocytopenia, or anemia) |
2. At least bilineage morphologic myelodysplasia |
3. Acquired clonal cytogenetic abnormality in hematopoietic cells |
4. Increased blasts (≧5%) |
4.2.4 Morphology and Histology
BM can be hypo-, normo-, or hypercellular in pediatric MDS. In children with low-grade MDS, cellularity was reduced in about half of patients [9, 10]. Since hematopoietic cells in hypocellular low-grade MDS are often distributed in a patchy pattern (Fig. 4.2a), information obtained from aspiration cytology is limited [22, 23]. Therefore, BM biopsies, which mirror the topography and cellularity of the local hematopoiesis, should be performed at least twice in order to diagnose hypoplastic BM disorders in children [2, 22, 23]. Patchy erythropoiesis with impaired maturation accompanied by sparsely distributed granulopoiesis, in an otherwise adipocytic BM is a characteristic of hypocellular low-grade MDS (Fig. 4.2b) [2, 22, 23]. Since megakaryocytes are markedly decreased or absent, immunohistochemistry is mandatory for the detection of micromegakaryocytes. A subset of children with low-grade MDS shows normo- or hypercellular BM, in which morphological and histological findings are reminiscent of advanced MDS, i.e., hematopoietic cells are diffusely distributed and dysplasia may be prominent (Fig. 4.2c, d).
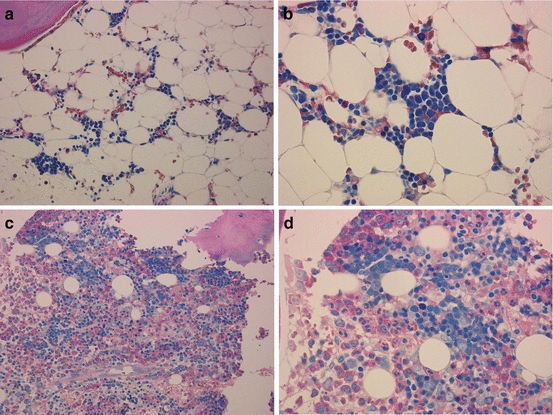
Fig. 4.2
Representative pictures of bone marrow (BM) biopsies obtained from patients with pediatric low-grade MDS (AS-D naphthol chloroacetate esterase stain with Giemsa staining), (a, b) show BM specimens of hypocellular refractory cytopenia of childhood, whereas (c, d) show those of hypercellular refractory cytopenia with multilineage dysplasia (c, d). (a) Patchy distribution of erythropoiesis accompanied by sparsely distributed granulopoiesis, in an otherwise adipocytic BM. (b) Immature erythroid precursors form one or several islands. (c) Immature erythropoiesis and granulopoiesis are distributed diffusely. (d) Left-shifted erythroid and myeloid cells are increased. These pictures were provided by the courtesy of Dr. Masafumi Ito
Both PB and BM show characteristic dysplastic features such as megaloblastic changes of erythroid cells, psudo-Pelger-Huet anomaly of granulocytes, and micromegakaryocytes (Table 4.3) [24, 25]. Although dysplasia is a pathognomonic feature of MDS, dysplastic feature constitutes only one aspect of the morphologic diagnosis and can be observed in various non-clonal disorders in children (Table 4.4).
Erythroid series | |
Nuclear lobulation | Presence of erythroblasts with lobulated nuclei (kidney-shaped, bilobulated, multilobulated, bizarre irregular nuclear profile) |
Multinuclearity | Two or more distinctly separated nuclei of the same or of different sizes |
Megaloblastoid changes | At least 1.5 times the size of a normal poly- or orthochromatic erythroblast with coarse condensation of chromatin and an increased nuclear-to-cytoplasmic ratio or orthochromatic erythroblasts with decreased nuclear-to-cytoplasmic ratio and at least double the size of a normal erythrocyte of the same maturational state |
Cytoplasmic granules or inclusions | Presence of granules or nuclear fragments that can be definitely differentiated from ribosomal RNA |
Myeloid series | |
Pseudo-Pelger-Huet anomaly | Mature granulocytes with either a centrally located round to ovoid nucleus (monolobated type) or two round nuclei of similar size connected by a slender chromatin bridge (bilobated type) |
Bizarre nuclear shape | Abnormal nuclear shape, including irregularly lobulated nuclei of segmented granulocytes with chromatin clumping or large twisted bands, large bands or metamyelocytes, multinuclearity (two distinctly separated neutrophilic bands or segmented nuclei) |
A- or hypogranularity | Abnormal, neutrophil or azurophil granules have to be markedly or completely absent, and the cytoplasm of mature neutrophilic granulocytes has to stain pale blue/gray or translucent in the Romanowsky–Giemsa stain. All maturation stages except blast cells should be affected. |
Nuclear/cytoplasmic (N/C) asynchrony | Mature neutrophilic granulocytes and metamyelocytes with basophilic cytoplasm and myelocytes with neutrophilic cytoplasm |
Megakaryocytic series | |
Micromegakaryocyte | Mononucleated megakaryocyte with a size comparable to that of a promyelocyte or less, lacking features of a blast cell |
Small binucleated megakaryocyte | Small megakaryocyte with the size of a micromegakaryocyte or slightly larger, with two round well-separated nuclei. |
Megakaryocyte with small round separated nuclei | Megakaryocytes of any size with multiple, at least three, round separated nuclei |
Megakaryocytes with nonlobated round nucleus | Megakaryocytes of normal or reduced size with a nonlobated round nucleus and a mature granular cytoplasm |
Table 4.4
Disorders with cytopenia, hypoplastic bone marrow, and dysplastic features
• Non-hematological disorders |
– Rheumatic disease |
– Congenital immunodeficiency (e.g., Wiskott-Aldrich syndrome) |
– Metabolic disorders (e.g., mevalonate kinase deficiency) |
– Pearson syndrome |
– Drug-induced (e.g., valproic acid) |
– Infection(e.g., cytomegalovirus, human parvovirus) |
– Nutritional disorder(e.g., deficiency of vitamin B12 or folate) |
• Hematological disorders |
– Aplastic anemia |
– Inherited bone marrow failure syndrome (e.g., Fanconi anemia, dyskeratosis congenita) |
– Paroxysmal nocturnal hemoglobinuria |
– Hemolytic anemia |
– Hemophagocytic lymphohistiocytosis |
4.2.5 Cytogenetics
The frequencies of abnormal karyotype vary according to the subtype. In low-grade MDS, two-thirds of patients have normal karyotype [10, 22]. Monosomy 7 and other chromosomal abnormalities including trisomy 8 were seen in 10% and 10–20% of patients with low-grade MDS, respectively [10, 22].
In contrast, only a third of patients with advanced MDS have normal karyotype, and two-thirds have chromosomal aberrations, in which monosomy 7 is the most common and accounts for 20–30% of patients [12–14, 26]. The outcome for patients with monosomy 7 is not worse than that of other children with MDS [17, 26]. Structural complex abnormalities defined as ≧3 chromosomal aberrations including at least one structural aberration are associated with a very poor outcome [12, 26]. Favorable cytogenetic aberrations defined by the prognostic scoring system in adult MDS such as -Y, del(11q), del(20q), and del(5q) [27] are rarely seen in children, and their prognostic significances remain to be established in pediatric MDS [17].
4.2.6 Pathobiology
Intrinsic defects in hematopoietic stem cell caused by acquired cytogenetic and genetic abnormalities are thought as hallmark features of adult MDS. Recently, molecular basis has been comprehensively addressed in a large cohort of MDS in adults [28]. About 90% of adult patients with MDS carry at least one oncogenic mutation, and two-thirds of them are found in individuals with a normal karyotype. Driver mutant genes in MDS include those of RNA splicing machinery (SF3B1, SRSF2, U2AF1, and ZRSR2), DNA methylation (TET2, DNMT3A, and IDH1/2), chromatin modification (ASXL1 and EZH2), transcription regulation (RUNX1), DNA repair (TP53), signal transduction (CBL, NRAS, and KRAS), and cohesin complex (STAG2) [25]. Although mutations in CBL, DNMT3A, CSF3R, CALR, and spliceosome genes have been reported to be rare in pediatric MDS so far [29–32], the landscape of molecular pathogenesis in pediatric MDS is still largely unknown. Epigenetic alterations such as hypermethylation of CDKN2B gene occur in children with MDS at a similar frequency to adults [33].
Considering that 10–25% of children with MDS have a known constitutional disorder [6, 7], inherited abnormalities predisposing to MDS/AML might play an important role in pathogenesis in pediatric MDS. Familial MDS/AML syndromes are resulted from deficiencies of the hematopoietic transcription factors CEBPA, RUNX1, and GATA2 [21]. Of these genes, germ line GATA2 mutations are found to occur in 7% of children with primary MDS [34]. Mutation carriers were older at diagnosis and more likely to present with monosomy 7 and advanced disease.
Given that immunosuppressive therapy (IST) is effective in some children with low-grade MDS as well as MDS in adults or AA [35–37], a part of these disorders might share a common pathogenesis, that is, T-cell–mediated inhibition of hematopoiesis. Indeed, T-cell oligoclonality was identified in 40% of RCC patients [38]. Minor paroxysmal nocturnal hemoglobinuria clones, which are thought as a predictor of favorable response to IST, were also detected in 41% of RCC patients [39]. These findings suggest that pathogenesis attributes to activated-T cells in a subset of children with low-grade MDS.
4.2.7 Differential Diagnosis
In children, cytopenias with decrease of BM cellularity might be caused by a various underlying disorders (Table 4.4) [22]. Of those, AA and IBMFS are the most common and important differential diagnoses of low-grade MDS; however, the clinical and histopathological distinction between these three disorders is challenging. Since IBMFS, such as FA and dyskeratosis congenita (DC), show overlapping morphological features with low-grade MDS and AA and children without any phenotypic features can be diagnosed with IBMFS, IBMFS have to be excluded by careful past medical and family history and thorough physical examination. In fact, FA and DC were found in patients with hypo- or normocellular RCC with a prevalence of 14% and 2%, respectively [40]. Several laboratory studies, such as chromosome breakage test and telomere length assay, should be considered in differential diagnosis of hypoplastic BM disorders.
AML is the major differential diagnosis of advanced MDS [24]. Blast percentage and morphological finding in a single specimen are insufficient to differentiate MDS from AML, and clinical features and cytogenetic findings should be taken into account in differential diagnosis. For example, monosomy 7 is suggestive of MDS, whereas significant organomegaly and hyperleukocytosis are suggestive of AML [13]. However, a small subset of children may have borderline features with BM blasts of 20–30% and no cytogenetic abnormalities, and reevaluation of the BM examination after 2 weeks is recommended.
4.2.8 Management and Treatment
Since MDS is clonal hematopoietic stem cell disorder and residual “healthy” stem cells are limited, hematopoietic cell transplantation (HCT) is the mainstay of management for children with MDS. Other therapeutic strategies such as hematopoietic growth factors, immunomodulatory drugs, or low-dose chemotherapy are adopted as clinical practice in adult MDS who are not candidate for HCT, but these approaches are generally not indicated in children. Azacitidine, a hypomethylating agent, has demonstrated clinical efficacy that prolongs survival in adult with MDS [41]. A retrospective analysis suggested that azacitidine was also safe and effective in some children with MDS [42]. However, because most children who benefited from azacitidine showed stable disease, the role of azacitidine in childhood MDS may be providing time for donor search before HCT or a less toxic option in palliative situations.
HCT for children with advanced MDS is a still great challenge because of high risk of relapse and transplant-related mortality (TRM). Myeloablative conditioning regimen consisting of busulfan, cyclophosphamide, and melphalan followed by HCT resulted in a 63% overall survival [12]. The 5-year cumulative incidence of TRM and relapse is 21% each. Age at HCT greater than 12 years, interval between diagnosis and HCT longer than 4 months, and occurrence of acute or extensive chronic graft-versus-host disease (GVHD) were associated with increased TRM, whereas more advanced disease was associated with the risk of relapse [12]. Treatment outcome of children with MDS underwent cord blood transplantation seemed suboptimal [43]. Because of lack of firm evidence due to paucity of patients, appropriate conditioning regimen, GVHD prophylaxis, and stem cell source for children with advanced MDS remain to be established. In addition, the significance of pre-transplant intensive chemotherapy also has yet to be elucidated. A Japanese study adopted induction therapy with etoposide, cytarabine, and mitoxantrone prior to HCT in 16 children with advanced MDS and found that complete remission rate was 81%, and no toxic deaths occurred; however, whether this strategy improved overall outcome was unclear [14]. A European study indicated that intensive chemotherapy before HCT did not show any difference in relapse, treatment-related mortality, or survival [12]. It is interesting to note that patients with very advanced MDS (blasts ≧30%) may benefit from pre-transplant intensive chemotherapy [12].
Clinical courses of low-grade MDS are heterogeneous: approximately a third of patient remained stable for long periods without IST or HCT, whereas a subset of patients suffered from progressive disease [10]. Monosomy 7 significantly correlated with disease progression and patients whose disease progressed before HCT fared significantly worse [9]; therefore, those with unfavorable karyotype such as monosomy 7 and complex karyotype are encouraged to be transplanted as soon as possible. Those who suffer from transfusion dependency or severe neutropenia may also be candidates for HCT if suitable donor is available. Because TRM is the major cause of treatment failure in children with low-grade MDS [44], reduced-intensity conditioning (RIC) regimen is an attractive option [45, 46]; however, it remains to be established whether RIC is an appropriate regimen for children with low-grade MDS with multilineage dysplasia, hypercellularity, or unfavorable karyotype.
If children with low-grade MDS without unfavorable karyotype require therapeutic intervention but suitable donor is unavailable, IST may be an advisable option. IST consisting of antithymocyte globulin (ATG) and cyclosporine has proven to be effective in 40 to 60% of children with low-grade MDS [10, 11, 36, 37, 47]. Some patients with chromosomal abnormalities or multilineage dysplasia were also reported to respond to IST [10, 11, 36, 37, 47]. However, patients who received IST remain at risk of clonal evolution and relapse, and the long-term failure-free survival rate was estimated as only 40–50% [10, 47]. As reliable biomarkers that can predict response have not yet identified, candidates for IST should be selected with utmost caution.
The outcome of children with therapy-related MDS is very poor. The survival of patients who receive only chemotherapy is dismal, and even HCT offers a cure only in 20–30% of patients [18, 48]. Compared with the outcomes of HCT in patients with therapy-related MDS, the results of secondary MDS/AML after AA appear to be favorable with a 41% overall survival rate [49]. Because TRM was the major cause of treatment failure, less toxic preparative regimens may be desirable in patients who develop secondary MDS/AML after AA. It is difficult to discuss the outcome of secondary MDS/AML arising from IBMFS because of its heterogeneity. Treatment with HCT in children with FA who have acute leukemia is challenging because of TRM and a high relapse rate. Data from the Center for International Blood and Marrow Transplant Research indicated that FA patients with MDS/AML had significantly worse 5-year survival than did patients with cytogenetic abnormalities alone (43% vs. 67%) [50]. In order to improve the outcome after HCT in children with FA and MDS/AML, low-dose chemotherapy prior to HCT or addition of high-dose cytarabine to fludarabine-based conditioning regimen has been attempted [51, 52].
4.3 Juvenile Myelomonocytic Leukemia
4.3.1 Epidemiology
JMML has an incidence of one in 1.2 million children per year and accounts for 3% of all hematologic malignancy in children [53]. The incidence of MDS and JMML in Japan was already described. The age at diagnosis is 2 years, ranging from 0.1 to 11, and males are predominating as a male to female ratio of 2:3 [4]. The reason why the disease affects more boys has not been elucidated.
4.3.2 Classification
The entity of JMM was established in 1997 [4]; however, diagnosis of JMML is not so simple, and a variety of clinical and laboratory findings may make diagnosis even more difficult. The clinical findings of JMML sometimes mimic human herpesvirus infections, leukocyte adhesion deficiency, infantile malignant osteopetrosis, hemophagocytic lymphohistiocytosis, and Wiskott-Aldrich syndrome [54]. Also, several important molecular findings have been identified, and they have been incorporated into the diagnostic criteria of JMML. Table 4.5 is a most recently issued diagnostic criteria proposed by Locatelli and Niemeyer [55], and it may be wise to realize that the criteria will be always changing in the future according to new findings to come.
I. Clinical and hematologic features (all 4 features mandatory) |
• Peripheral blood monocyte count >1 × 109/L |
• Blast percentage in peripheral blood and bone marrow <20% |
• Splenomegaly |
• Absence of Philadelphia chromosome (BCR/ABL rearrangement) |
II. Oncogenetic studies (1 finding is sufficient) |
• Somatic mutation in PTPN11 a or KRAS a or NRAS a,b |
• Clinical diagnosis of NF1 or germ line NF1 mutation |
• Germ line CBL mutation and loss of heterozygosity of CBL c |
III. Only for those patients (10% of the whole number) without any oncogenetic parameter, besides the clinical and hematologic features listed under I, at least 2 of the following criteria have to be fulfilled: |
• Monosomy 7 or any other chromosomal abnormality |
• HbF increased for age
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|