Introduction
Diabetes is a heterogeneous disorder with many different possible causes, both genetic and acquired. Risk for the most common causes, type 1 and type 2 diabetes, depends on many different gene loci with intermediate or low effects and are thus considered polygenic. However, approximately 1% to 5% of all diabetes is caused by abnormalities at a single gene or locus and as a group, these entities are termed monogenic diabetes. Over 30 genes have now been described as causing the various forms of monogenic diabetes, where they will typically fall into one or more of the three main overlapping phenotypic categories: neonatal diabetes, maturity-onset diabetes of the young (MODY), and syndromic diabetes. In this chapter, we describe several of the more common causes of neonatal diabetes in some detail, and also provide brief descriptions of some of the main forms of MODY and syndromic diabetes, with a focus on diagnosis and treatment.
Definition
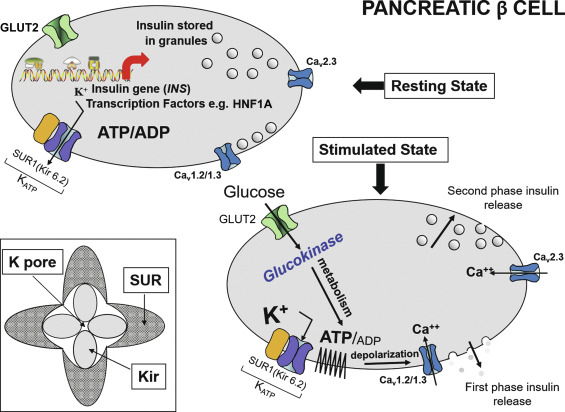
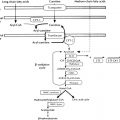
Neonatal diabetes mellitus (NDM) was the initial term used to describe the presentation of diabetes within the first few days or weeks of life and was defined further in early reports as severe hyperglycemia occurring in the first month of life, lasting at least 2 weeks, and requiring insulin therapy to control blood glucose. These strict criteria have been progressively loosened as it became evident that the exact age at which the diabetes was diagnosed was more variable, even when an underlying cause, distinct from autoimmune type 1 diabetes mellitus (T1DM), was suspected, such as a genetic cause for pancreatic malformation, faulty insulin synthesis or secretion. Accumulated evidence has suggested an age under 6 months as being a particularly likely indication of NDM, because the majority of cases with an underlying monogenic cause will be diagnosed under 6 months, whereas autoimmune T1DM is highly unlikely during this window. Increasingly, however, it has become recognized that, for reasons that are poorly understood, several of the genetic forms can initially present with diabetes as late as 9 months to 1 year or even later. Indeed, pedigree analyses conclusively demonstrate that the same defect that causes permanent or transient NDM can be present in parents or other first-degree relatives with a phenotype consistent with MODY, but may have been misdiagnosed as T1DM or type 2 diabetes mellitus (T2DM) as subsequently detailed. Herein lies the importance of understanding the genetic basis of NDM, for although these entities are rare, they have taught us much about the genetic pathways involved in the formation of the exocrine and endocrine pancreas. For example, it has been shown that the specific combination of three transcription factors, Ngn3, Pdx1 , and Mafa , known to be implicated in the determination of cell lineage during pancreas formation, can reprogram adult mouse exocrine pancreatic cells into cells that closely resemble pancreatic beta cells. Such information is essential for the ultimate ability to generate beta cells and whole islets as potential therapies for any form of diabetes, including T1DM. Equally important is the demonstration that activating mutations in the pore-forming potassium inward rectifying channel, family 6 subtype2- KCNJ11 gene (Kir6.2) and its regulatory subunit sulfonylurea receptor 1- ABCC8 gene (SUR1) of the K ATP -regulated potassium channel, which keep the channel open and hence limit or preclude insulin secretion resulting in NDM ( Fig. 10.1 ), can be overcome by high-dose sulfonylurea therapy, which restores endogenous insulin secretion in response to feeding. Because of the restoration of endogenous insulin secretion that can be triggered by the incretin effect in response to feeding, this oral treatment provides better metabolic control than multiple daily injections of insulin or insulin pumps and with a better quality of life. Successful transfer to sulfonylureas is best predicted by in vitro response of specific mutation and diabetes duration. These findings emphasize the benefits of research for understanding pathophysiology and choosing appropriate treatment. Indeed, for those whose NDM is caused by mutations in the K ATP channel that respond to sulfonylureas, these treatments border on the miraculous.
Incidence
Early estimates placed the incidence of NDM at approximately one in 500,000, but as increased awareness has led to these entities being recognized more frequently, the reported incidence has risen considerably. In populations with high rates of consanguinity, some studies report an incidence of NDM as high as one in 21,000 births. A large representative database for pediatric diabetes reported that the incidence of NDM represented approximately one case in 89,000 live births in Germany, and a similar incidence occurred in Italy. In three other European countries, the incidence was reported to be one in 260,000 live births only for those with permanent NDM (PNDM) (suggesting a higher incidence if transient forms of NDM [TNDM] were also included). In the SEARCH for Diabetes in Youth Study involving 15,829 subjects aged under 20 years diagnosed with diabetes during the years 2001 to 2008, 39 were diagnosed before the age of 6 months. Among these 39 subjects with onset less than 6 months of age, 35 had permanent neonatal diabetes and an additional three had TNDM, leaving one subject whose status remained unknown. Hence, the total prevalence among children diagnosed with diabetes was approximately 0.246% or approximately 1 in 400. The majority were classified by their primary care providers as having T1DM and treated with insulin; only seven underwent mutational analysis for three of the most common genes ( KCNJ11, ABCC8 , and the insulin gene [ INS] ) and five of these seven had mutations in one of these three genes. The estimated population prevalence of PNDM in those aged less than 20 years in that study was one in 246,000 people.
Clinical presentation
Infants affected with diabetes may have diabetes in isolation, or the underlying genetic defect may also cause a variety of other clinical features ( Fig. 10.2 ). Infants with NDM are more likely to be born small for gestational age, and in many cases there may also have been concern for intrauterine growth retardation (IUGR) during their pregnancy, reflecting the in utero deficiency of insulin and emphasizing the role of insulin as a determinant of fetal growth. Their small size and low birth weight markedly contrasts with the large birth weight and size of infants with inactivating mutations in the same K ATP genes that instead of diabetes lead to hyperinsulinemic hypoglycemia (see Chapters 7 and 23 ). A disproportionate number of those with NDM are born prematurely at less than 37 weeks’ gestation, which in some cases may be associated with induction of the delivery because of their IUGR status. Hyperglycemia leads to osmotic diuresis and avid feeding from breast or bottle despite which the infants often fail to thrive. Delay in diagnosis from not considering the possibility of diabetes mellitus in a newborn may lead to severe dehydration and life-threatening diabetic ketoacidosis (DKA); in fact, a recent retrospective study of a large series of NDM cases in the United States showed that 66% of patients presented in DKA. Rare syndromic forms may have severe congenital malformations, such as intestinal and biliary atresia ( RFX6 ), congenital heart defects ( GATA6 or GATA4 ), or brain malformations ( PTF1A , NEUROD1 , IER3IP1, MNX1, NKX2.2 ), or alternatively may have other features, such as skeletal dysplasia ( EIF2AK3 ) that are difficult to recognize during the neonatal period. In addition to mutations in FOXP3 responsible for the IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked), several additional genes ( STAT1, STAT3 , LRBA , IL2RA ) have now been described to have mutations causing infancy-onset diabetes, along with other autoimmune disorders that result from dysfunction of immune regulation.
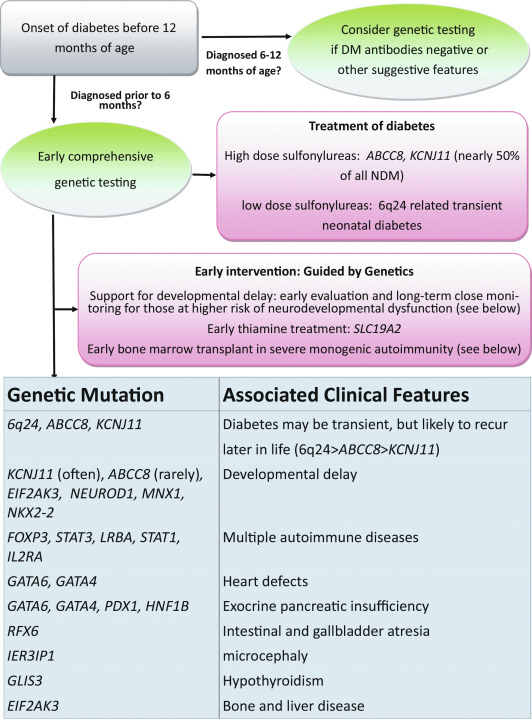
Patients with overexpression of genes at chromosome 6q24 present with TNDM and will often have more subtle features, such as macroglossia and umbilical hernia, reminiscent of Beckwith-Wiedemann syndrome. In addition, they rarely may have dysmorphic facies, as well as renal tract anomalies, such as hydronephrosis and vesicoureteral reflux, a variety of cardiac anomalies, hypothyroidism, or hand-finger anomalies. A coarse facial appearance together with epilepsy and later manifestations of developmental delay constitute the developmental delay, epilepsy, neonatal diabetes (DEND) syndrome associated with the most severe mutations of the KCNJ11 gene ; however, in most cases of KCNJ11 or ABCC8 mutations that maintain the K ATP channel in a variable open state and hence limit insulin secretion, there will not be any abnormal findings on physical examination. Even in those with the severe mutations causing significant neurodevelopmental dysfunction, the abnormalities may often not be recognized until patients are older. Clinical manifestations involving organs other than the pancreas may be part of the syndromes of a variety of genetic causes of NDM. These features in patients with NDM were previously incorporated in a retrospective categorization system to describe permanent, transient, and syndromic forms of NDM. However, now that the monogenic basis of NDM has been well established, genetic testing should be done immediately upon diagnosis of diabetes in the neonatal/infancy period, at which time it will be unclear whether the baby has a permanent or transient form of NDM (see Fig. 10.2 ). Furthermore, the extrapancreatic manifestations of genes causing syndromic NDM can be phenotypically quite variable and many of these features will often not be readily apparent during the neonatal period. Expert consensus has therefore shifted toward obtaining comprehensive genetic testing utilizing next-generation sequencing (NGS) panels (or a tiered approach) rather than iterative testing of fewer genes selected based on clinical features.
Transient neonatal diabetes mellitus
TNDM is so named because hyperglycemia resolves spontaneously within the first few months of life and no longer requires treatment, although it may reappear later in life. About 70% of these cases will have TNDM1 (6q24-related TNDM), whereas the majority of the remaining cases of TNDM will be caused by mildly activating mutations in the K ATP genes ABCC8 (SUR1) and KCNJ11 (Kir6.2), and only a small minority will be caused by recessive insulin gene mutations, mutations in the transcription factor HNF1B , or mutations in SLCA2A , the gene encoding the GLUT2 transporter.
6q24-Related Transient Neonatal Diabetes Mellitus (TNDM1)
TNDM1 (or 6q24-related TNDM) is caused by the overexpression of genes at the 6q24 chromosomal locus, including pleomorphic adenoma gene-like 1 ( PLAGL1 ), which is a proapoptotic zinc finger protein, and (hydatidiform mole-associated and imprinted transcript ( HYMAI ), encoding an untranslated messenger ribonucleic acid (mRNA). TNDM1 arises as a result of uniparental paternal disomy of the entire—or only a segment that includes 6q24—chromosome 6 (UPD6), a paternally inherited duplication of chromosome 6q24, or relaxation of imprinting of the maternally methylated genes on chromosome 6 ( Fig. 10.3 ). It is important to emphasize that these chromosomal changes cause altered expression of genes, rather than representing mutations. For example, PLAGL1 ( ZAC ) has antiproliferative properties and is thought to function as a tumor growth suppressor expressed only on the paternal allele ; overexpression in fetal life is believed to lead to underdevelopment of the pancreas. Although the precise mechanisms by which overexpression of PLAGL1 lead to TNDM1 are not known, overexpression of ZAC in a clonal pancreatic beta cell line impairs glucose-stimulated insulin translation and secretion. A transgenic mouse model expressing the human TNDM1 locus (6q24) is characterized by impaired glucose homeostasis with hyperglycemia in the neonatal period and impaired glucose tolerance with reduced insulin responses to intravenous (IV) glucose as adults. The pancreata of these animals display reduced expression of endocrine differentiation factors, notably PDX1, NGN3, and PAX6. There is also a reduction in the number of insulin staining cells and reduced insulin content or insulin secretion despite normal or elevated beta cell mass at all postnatal periods. Thus this model recapitulates TNDM1 and suggests that altered expression of ZAC/HYMAI cause impaired development of the endocrine pancreas, as well as impaired beta cell function. This mouse model also demonstrates resolution of abnormal insulin secretion with restoration of normal glucose tolerance during the “juvenile” phase of mouse development between 1.5 and 2 months of life, during which there is an approximate doubling of beta cell number that compensates for the reduced insulin synthesis and secretion of each cell. Also as in humans, the compensatory increase in beta cell mass is not sustained, resulting in a mild diabetes mellitus characterized by normal fasting glucose but hyperglycemia after glucose challenge. Overall, despite the recapitulation of the key features of the human disease, the mouse model displays milder features. One possible reason for this milder phenotype in mice is that pancreatic expression of the mouse ortholog Zac1 declines drastically during gestation and early postnatal growth in mice, whereas expression of the ZAC gene in human pancreas declines between the second trimester and adult life. More important, ZAC was specifically expressed only in the islets of the human fetus, whereas Zac1 was predominantly expressed in mesenchyme of the mouse embryo, which may explain the milder features in the mouse model of TNDM1.
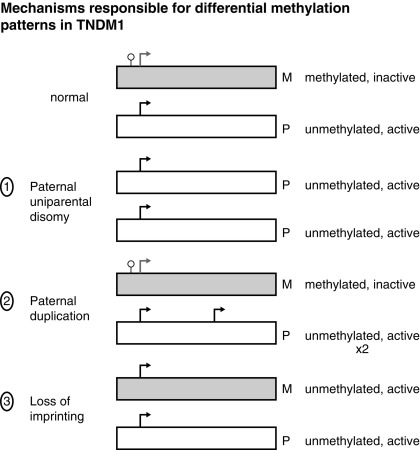
In patients with TNDM1 resulting from hypomethylation of the maternal differentially methylated region (DMR) of chromosome 6q24, there may be hypomethylation of other maternally imprinted loci (HIL) throughout the genome. In cases displaying a more generalized HIL, the majority have a mutation in the transcription factor zinc finger protein 57 ( ZFP57 ). HIL also occurs in Beckwith-Wiedemann syndrome and this likely explains the macroglossia, umbilical hernia, and several congenital abnormalities described in TNDM1. In a large multinational cohort involving 163 patients with TNDM1, the authors describe IUGR with a mean birth weight of 2001 ± 417 g (mean ± standard deviation) and adjusted Z score for birth weight of − 2.5. The mean age of presentation was 8 ± 12 days with a median of 4 days and a mode of 1 day. Mean gestation was 37.8 ± 2.4 weeks and prematurity was significantly more common than in the general population. Remission occurred at a mean age of 4.5 ± 5.8 months with a median at 3 months. Age at presentation was positively correlated to gestational age, but age at remission was negatively correlated with adjusted birth weight. Thus the higher the birth weight, the earlier the remission and vice versa. This would be consistent with the effects of insulin on intrauterine growth, so that the larger infants would have the milder defect and therefore tend to enter remission sooner. Congenital anomalies were significantly more frequent in patients with UPD6 or HIL. Hypomethylation defects were overrepresented in patients born after assisted conception. Thus babies with TNDM1 generally present with diabetes mellitus within the first days of life, are small, and may have been born prematurely. The presence of one or more congenital anomalies suggests UPD or multiple HIL, and among the latter, almost one in seven had been conceived with assisted reproductive techniques. Macroglossia is present in about 50%, umbilical hernia in about 25%, and facial dysmorphism in about 20%. Cardiac and renal anomalies (~ 9%), hand abnormalities (~ 8%), and hypothyroidism (~ 4%) also may be present. Remission, when it occurs, is usually around 3 months and about half of these patients will revert to varying degrees of hyperglycemia in the teen years or later. An unusual manifestation following remission of diabetes in patients with 6q24 methylation defects is hypoglycemia with hyperinsulinemia, most often when the cause is UPD6. Modern molecular techniques permit diagnosis to be established which then influences treatment; however, many commercial sequencing panels for monogenic diabetes do not include testing that will reveal overexpression of genes at 6q24 as a cause of diabetes.
Patients with TNDM1 are sensitive to insulin and respond with excellent catch-up growth within several weeks of treatment. Progressive reduction of the insulin dose required to control blood glucose while avoiding hypoglycemia heralds the onset of remission. Although these patients do well with insulin treatment, some reports suggest a variable response to oral sulfonylurea treatment; though it is difficult to discern the extent to which the clinical improvement was caused by medical treatment versus spontaneous resolution of the condition in these observational studies. Of note, rare cases with 6q24-related TNDM will develop significant hypoglycemia within weeks or months after the remission of hyperglycemia. Most had evidence for hyperinsulinism as the cause of hypoglycemia and responded well to diazoxide treatment that was still required after several years in some cases. Even after resolution of diabetes during infancy, it is important that these patients understand that diabetes is highly likely to recur around the time of adolescence. Although a few papers report good response to other drugs besides insulin, the best approach to monitoring during remission, as well as to treatment after recurrence, remain uncertain. Because many questions remain about this condition, it is important that such cases be referred to research centers tracking long-term outcome, even if the diabetes has gone into remission.
K ATP Channel-Related Transient Neonatal Diabetes Mellitus (TNDM2)
TNDM2 is one classification given to diabetes that remits during infancy and may recur later in life but is caused by mutations in genes regulating insulin secretion rather than expression of imprinted genes. The majority of these entities are caused by activating mutations in the K ATP channel genes ABCC8 and KCNJ11 , which, respectively, code for the SUR 1 and Kir6.2 subunits of the K ATP channel (see Fig. 10.1 ). Recessive loss-of-function mutations in the INS itself may occasionally be responsible for TNDM2 ; the autosomal dominant insulin gene mutations are associated with PNDM. There are rare reports of HNF1B and SLC2A2 mutations also associated with TNDM.
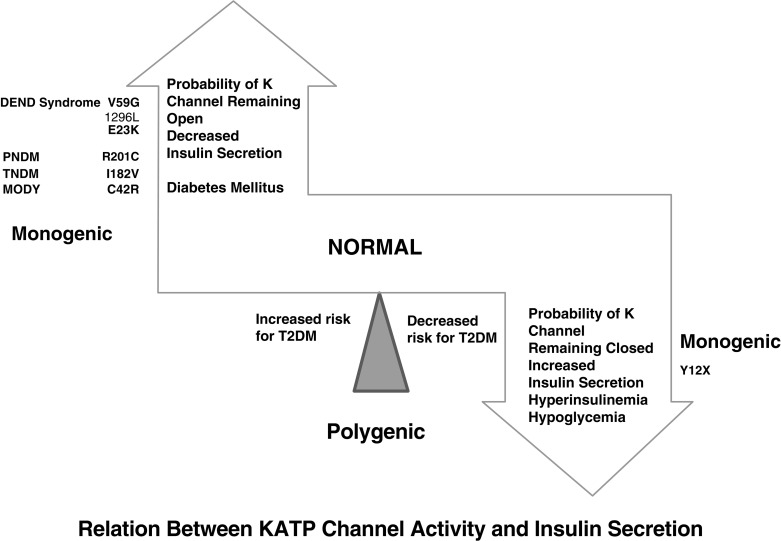
The normal state of the K ATP channel is to remain open, setting the resting membrane potential of the beta cell. Insulin secretion occurs when the channel closes in response to an increase in adenosine triphosphate (ATP) generated from the metabolism of glucose or amino acids, thereby changing the ATP:adenosine diphosphate (ADP) ratio. Closure of the channel with intracellular retention of K + causes depolarization of the plasma membrane, opening of voltage-gated calcium channels, influx of calcium, and secretion of insulin. Activating mutations in ABCC8 or KCNJ11 alter the ability of the channel to respond to the change in the ATP:ADP, so that the channel remains open to some extent, efflux of K + from the beta cell continues, permitting the cell membrane to remain hyperpolarized and therefore resulting in various degrees of impaired insulin secretion. These same mechanisms are also responsible for the most common form of PNDM, as subsequently discussed. It remains unclear how or why remission occurs, but it has been shown in vitro that mutations causing TNDM have a less pronounced effect on channel function compared with mutations that cause PNDM. The ability of ATP to close the channel in vitro correlates with the severity of NDM, including the severe permanent form associated with the DEND syndrome, which demonstrates the greatest resistance to closure by ATP in vitro. Both resistance to closure caused by activating mutations, or resistance to opening of the channel because of inactivating mutations, segregate with certain mutations as illustrated in Fig. 10.4 . As shown in the figure, near the fulcrum of this spectrum, those with minor defects may be prone to either develop milder type 2 diabetes or may be resistant to the development of diabetes by virtue of enhanced insulin secretion (see Fig. 10.4 ).
In comparison with TNDM1, patients with TNDM2 generally have greater birth weight, present or are diagnosed with diabetes mellitus later, remit later, and recur earlier ( Table 10.1 ). Family members of patients with these forms of TNDM2 may have diabetes that was diagnosed in adulthood and was considered to be T1DM, T2DM, or MODY and yet harbor the same heterozygous mutations as the proband with NDM. This reflects the variable penetrance of these genes or upstream factors that may modify the expression of the gene in different individuals. Confirming the presence of an ABCC8 or KCNJ11 mutation in a case of NDM is important for management because most of these K ATP channel mutations respond to sulfonylurea treatment both at the time of initial diagnosis or later at the time of relapse. During their remission phase, these patients do not require therapy.
| Characteristic (median) | ABCC8 / KCNJ11 (n = 25) | 6q24 * (n = 23) | P-Value |
|---|---|---|---|
| Age at diagnosis (weeks) | 4 (0–16) | 0 (0–4) | < .001 |
| Age at remission (weeks) | 35 (2–208) | 13 (5–60) | < .001 |
| Age at relapse (years) | 4.7 (3–15) | 16 (4–25) | .073 |
| Birth weight (g) | 2570 (1360–3570) | 1950 (1600–2670) | < .001 |
| Percentile birth weight | 12 (< 1st–89th) | < 1st (< 1st–21st) | < .001 |
* Data previously reported (Temple IK, Gardner RJ, Mackay DJ, Barber JC, Robinson DO, Shield JP: Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes 49: 1359–1366, 2000). Differences between groups were calculated using Mann-Whitney U and χ2 tests. Centile birth weights were calculated according to U.K. growth charts (Cole TJ, Freeman JV, Preece MA: British 1990 growth reference centiles for weight, height, body mass index and head circumference fitted by maximum penalized likelihood. Stat Med 17: 407–429, 1998) because the majority of patients were of U.K. white origin.
Recessive loss of function mutation in the INS gene has been reported in rare patients with TNDM. These patients appear to enter remission at a median age of 12 weeks and insulin was required before remission and after later relapse that was reported in some cases.
Patients with mutations in the transcription factor hepatocyte nuclear factor 1 beta ( HNF1B ) are known to have diabetes associated with renal cysts with onset at a median of 20 years. However, two reported patients had neonatal diabetes: one was diagnosed at age 15 days and required insulin, initially intermittently and then permanently; a second patient diagnosed at the age of 17 days had remission 2 weeks after diagnosis but relapsed at the age of 8 years. There have also been a few reported cases of mutations in SLC2A2 , which encodes the glucose transporter type 2 (GLUT2). Recessive inactivating mutations in this gene cause Fanconi-Bickel syndrome, characterized by glucosuria, galactosuria, aminoaciduria, proteinuria, and phosphaturia, as well as rickets, poor growth, and short stature with associated glucose and galactose intolerance and enlarged livers. Transient neonatal diabetes has been reported in these patients to occur in association with the classic Fanconi-Bickel syndrome.
Permanent neonatal diabetes mellitus
The majority of genes that cause NDM result in PNDM without any significant treatment remission. Approximately 80% to 85% of infants with diabetes diagnosed before 6 months of age will have an underlying monogenic cause that can be identified, and the remainder may carry as-yet uncharacterized defects. Although diabetes resulting from monogenic causes can also be diagnosed between 6 and 12 months of age or later, and genetic testing can be considered, the majority of diabetes cases in this age range will have autoimmune diabetes (T1D) that can be suggested by positive anti-beta cell autoantibody testing or type 1 diabetes genetic risk scores (routinely done in most NDM research centers). Except in certain populations with higher frequency of consanguineous births, the vast majority of PNDM is caused by mutations in three genes: the two K ATP channel genes KCNJ11 coding for the pore-forming protein Kir6.2 (50%) and ABCC8 coding for SUR1 (together account for 40%–45% of cases; see Figs. 10.1 and 10.2 ), and mutations in the gene encoding insulin itself ( INS ). Dominant mutations in GATA6 or GATA4 causing pancreatic hypoplasia are rarer than other dominant causes, as are X-linked mutations in FOXP3 causing the IPEX syndrome, or milder IPEX-like syndromes, whereas the remaining gene causes of PNDM are very rare recessive disorders that often include a variety of extrapancreatic syndromic features (see Fig. 10.2 ).
K ATP Mutations: KCNJ11 and ABCC8
Until the discovery of the genes responsible for the K ATP channel, patients with PNDM were considered to have insulin-dependent diabetes mellitus; now it is known that the majority of patients with K ATP mutations causing PNDM respond to oral therapy with sulfonylurea. Early genetic testing is imperative because those who are responsive to sulfonylurea treatment and have earlier initiation of treatment have been reported to have improved response to therapy and may help improve quality of life. Typically treated with high-dose sulfonylureas, a cohort with this treatment has shown mild and infrequent hypoglycemia. Patients with PNDM caused by mutations in the K ATP genes or the insulin gene usually present at 2 to 3 months of life, and sometimes later, and may be in severe DKA by the time of the diagnosis. Activating mutations in the Kir6.2 subunit of the K ATP channel were first reported in 2004, being found in 10 out of 29 subjects with PNDM. Whereas the insulin secretory response to IV glucagon and glucose was minimal, there was a better response to oral glucose. Patients did respond with insulin secretion to IV tolbutamide, clearly hinting at possible therapy by the administration of sulfonylurea and confirmed by a landmark study published 2 years later. Expression of the mutated Kir6.2 subunit together with a normal SUR1 subunit in Xenopus laevis oocytes revealed that the ability to enable channel closure by ATP was greatly impaired. This provided a means to correlate the degree of in vitro abnormality with the clinical severity of the diabetes. The affected patients predominantly had de novo mutations, with only 20% of the mutations inherited from a parent. It was also noted that 4 of 10 of the patients had severe developmental delay, muscle weakness, and epilepsy, as well as dysmorphic facial features, which was termed the DEND syndrome . The degree of muscle weakness was partially ameliorated by treatment with sulfonylurea, raising the possibility that the developmental delay and epilepsy may also be ameliorated, or perhaps prevented, by early recognition and treatment with sulfonylurea. Subsequently, it was demonstrated in mice that transgenic expression of an activating mutation in Kir6.2 in mouse pancreatic beta cells recapitulates neonatal diabetes and that the muscle dysfunction caused by a human K ATP channel mutation is neuronal and not muscular in origin. K ATP channels exist in other tissues and are known to modulate electric activity and neurotransmitter release at brain synapses in various regions of the brain. Moreover, K ATP channels in the ventromedial hypothalamic nucleus may be involved in the counterregulatory response to hypoglycemia ; in the arcuate nucleus neurons, K ATP channels may be involved in appetite regulation.
A spectrum of clinical disturbances occurs with different mutations ranging from the DEND syndrome, to relapsing diabetes, permanent diabetes appearing initially in childhood or later in adults. Mutations in adjacent locations may cause either neonatal diabetes or hyperinsulinism because they increase or decrease the open state of the channel. Likewise, mutations in the ABCC8 gene encoding SUR1 cause transient or permanent NDM, or permanent diabetes diagnosed beyond the newborn period in children, or in adults, and mutations at a similar site in the gene can result in either hyperinsulinism or neonatal diabetes.
Initial reports noted certain mutations (such as KCNJ11 V59M) are characterized by significant global developmental delay (often termed intermediate DEND syndrome , or iDEND ) in about 20% of cases, and anecdotal reports suggested improvement of neurological symptoms of weakness, dyscoordination, and visuomotor impairment after treatment with sulfonylureas (replacing insulin) was initiated. Several groups have since undertaken careful characterizations of large groups of patients using standardized measures that reveal a wide range of dysfunction, including behavioral difficulties and attention deficit hyperactivity disorder symptoms or diagnosis, lower educational attainment, and difficulties with executive functioning. In one study of 14 patients with mutations previously thought to cause diabetes in isolation, who did not have global developmental delay, IQ was found to be close to normal (91.1 ± 11.3) but significantly lower than 20 sibling controls (111.0 ± 8.3). Raising the importance of early proper diagnosis and treatment of these conditions, another study revealed better performance on a standardized measure of visuomotor functioning by the three patients who had been started on sulfonylurea treatment under a year, among eight patients with of V59M mutations in KCNJ11.
The safety and tolerability of high-dose sulfonylurea treatment, as well as its durability over more than 10 years of treatment, has also been reported in a few recent studies. A survey of 30 patients over a total of 166 patient years revealed no episodes of severe hypoglycemia, whereas mild to moderate hypoglycemia was unrelated to sulfonylurea dose. A recent follow-up study on 81 patients from a European cohort started on sulfonylureas before 2006 showed no episodes of severe hypoglycemia over 809 patient-years, with sustained excellent glycemic control on stable doses with infrequent mild side effects.
Insulin Gene Mutations
Insulin gene mutations as a cause of PNDM were first reported in 2007 and are now known to be the second most common mutations responsible for these entities. Inheritance was autosomal dominant in the familial cases, but the majority had de novo mutations. The mutations occurred in a critical region of the preproinsulin molecule, predicting misfolding and hence loss of normal trafficking of the proinsulin in the insulin secretory pathway. This misfolding was also proposed to induce the unfolded protein response, with degradation in the endoplasmic reticulum (ER), leading to severe ER stress and apoptosis of the beta cells, processes known to occur in mouse models of dominant insulin gene mutations. Clinically, the age at diagnosis averaged 13 weeks compared with 5 weeks for KCNJ11 and 7 weeks for ABCC8 mutations. With a normal range of gestational ages of 36 to 41 weeks, mean birth weight was also normal at 2846 g. Thus these abnormalities appear to disturb intrauterine growth less than in 6q24 TNDM or those with K ATP channel mutations. These dominant or de novo mutations are not usually associated with TNDM or remission, whereas recessive mutations of the insulin gene can result in a remitting type of NDM as described earlier. The initial suggestion of ER stress as a mechanism has been largely confirmed and the spectrum of disorders in the insulin gene extends to a MODY phenotype or onset in adulthood. Treatment of these patients is currently limited to insulin, whereby they can be managed very similarly to a patient with autoimmune T1DM. A few case series have demonstrated the effectiveness of insulin pumps and continuous glucose monitors in achieving good glycemic control as early as the neonatal period following diabetes diagnosis. One case study of two sisters suggested the theoretical benefit of early optimization of glycemic control to minimize the need for endogenous beta cells to respond to hyperglycemic excursions by producing mutated insulin that is toxic to beta cells. Limiting the toxic effects of protein production appeared to promote improved cell survival that might allow for at least low level production of insulin via the normal allele.
Other Genetic Forms of Permanent Neonatal Diabetes Mellitus
Two genes that cause MODY, the enzymatic metabolic gatekeeper glucokinase ( GCK ), and the islet formation transcription factor PDX1 (previously called IPF-1 ), cause monogenic forms of diabetes, respectively, known as MODY2 and MODY4 , when in the heterozygous state and can more rarely cause PNDM when in the homozygous or compound heterozygous state.
Glucokinase
Glucokinase (GCK) has been called the glucose sensor of the beta cell. It phosphorylates glucose to glucose-6-phosphate, permitting entry into the glycolytic pathway for metabolism and generation of ATP, which triggers insulin secretion (see Fig. 10.1 ). GCK is also known as hexokinase IV or hexokinase D and is most active in the physiological range of glucose at 4 to 10 mmol/L (72–180 mg/dL) with a K m of ~ 8 mmol /L (144 mg/dL). The glucose-stimulated insulin release (GSIR) threshold occurs when glucose phosphorylating capacity reaches 30% of maximum, which generally occurs under normal circumstances at a glucose concentration of ~ 90 mg/dL and is maximum at glucose concentrations of 300 mg/dL or higher but not reaching more than ~ 80% of full (100%) glucose phosphorylating capacity.
Activating mutations of GCK exert these effects at lower glucose concentrations and hence cause hyperinsulinemic hypoglycemia (see Chapters 7 and 23 ). Inactivating heterozygous mutations in GCK cause a mild diabetes because the glucose phosphorylating capacity is shifted to the right, so that the 30% phosphorylating capacity needed for the GSIR occurs when glucose concentrations are around 120 mg/dL rather than 90 mg/dL and reach only a peak of 50% to 60% of the 100% phosphorylating capacity. This is enough to clear all glucose after a meal, but with periods of hyperglycemia before this occurs. GCK inactivating mutations are one of the most common forms of MODY (see later), often presenting asymptomatically as an incidental finding discovered during blood tests done for other reasons, and there will often be a positive family history of diabetes.
Homozygous inactivating mutations in GCK were reported in two patients in 2001, and the same authors reported an additional three cases in 2003. The affected subjects were homozygous for the mutation or were compound heterozygous for two different mutations, being a splice site in one and a missense mutation in the other. Each of these five initial subjects was characterized by IUGR, permanent neonatal diabetes from day 1 of life, and hyperglycemia in parents, which was an important diagnostic clue. Apart from some other novel single case reports, an additional four novel cases of PNDM caused by homozygous mutations in GCK were reported in 2011. Overall, these mutations are rare in NDM; in one large cohort of 54 cases of NDM, GCK mutations were found in only one case of European ancestry, and the same was true for a study comparing the etiologies of PNDM in an Arab versus European population.
Pancreatic Duodenal Homeobox 1
Pancreatic duodenal homeobox 1 ( PDX1 ), previously known as insulin promoter factor 1 ( IPF1 ), is a critical transcription factor that defines the pancreatic progenitor cell as it differentiates from the endodermal epithelium. Exocrine-duct lineage is then specified by the pancreas transcription factor 1 (PTF1), which defines the exocrine and duct cells. However, in the presence of NGN3, the progenitor cell differentiates into a series of endocrine pancreatic cells under the influence of other transcription factors, including PAX4. Thus PDX1 is critical for the formation of both the exocrine and endocrine pancreas, and hence homozygous mutations will lead to an absence of pancreas formation with manifestations of neonatal diabetes and pancreatic exocrine insufficiency. The parents may be heterozygous carriers and therefore known to have a form of diabetes known as MODY4 . The first case of pancreatic agenesis from a monogenic cause in 1997 revealed the etiology as a homozygous loss-of-function frameshift mutation in PDX1 , whereas the second case was reported in 2003, and the third case was reported in 2009 ; each of these patients were characterized by pancreatic agenesis with manifestations of both exocrine and endocrine abnormalities. In a more recent study, 103 patients in whom K ATP and INS mutations had been excluded and who were not known to have abnormal development or clinical exocrine insufficiency of the pancreas, were tested and found to include four cases with homozygous or compound heterozygous PDX1 mutations; thus it is possible to have PNDM caused by a mutation in PDX1 without necessarily having complete pancreatic agenesis. In this regard, it is important to note that neonatal diabetes may be associated with pancreatic, as well as cerebellar, agenesis as a result of mutations in the PTF1A gene (pancreas transcription factor 1A) and that the NEUROG3 gene (known as Ngn3 in mice) can be associated with neonatal diabetes and congenital malabsorptive diarrhea consistent with the role of this gene in pancreas development as briefly described earlier. Other genes that have been implicated in pancreatic agenesis or hypoplasia include PTF1A , NEUROD1 , EIF2AK3 , HNF1B , RFX6 , GATA4, and GATA6 , which may also be associated with other features, such as congenital heart defects or cerebellar agenesis. As a diagnostic clue, if pancreatic imaging shows the absence or hypoplasia of a pancreas, the diagnostic search can be restricted to a series of genes as outlined.
Syndromic Neonatal Diabetes Mellitus
The syndromes associated with NDM together constitute a relatively small fraction of NDM that can vary considerably depending on the frequency of consanguineous unions in the population being studied. Although each form is rare, they are of interest in identifying the spectrum of genetic disorders that can result in abnormal pancreas formation, malfunction, or destruction. It is worth noting that the term syndromic is somewhat subjective but in this context helps to categorize very rare, usually recessive forms of PNDM. Even the most common forms of PNDM caused by K ATP mutations could be considered syndromic based on their frequent variable neurodevelopmental dysfunction, especially with the most severe mutations causing DEND syndrome. In the past, syndromic features could be used to point to specific genes to be sequenced, but consensus opinion now suggests that all NDM patients should undergo comprehensive genetic testing for all known causes either all at once as part of an NGS panel or by using a tiered approach.
EIF2AK3 : Wollcott-Rallison Syndrome
This rare condition is actually the most common genetic cause of PNDM in consanguineous families, and it may present as isolated nonautoimmune NDM diagnosed at 3 weeks of age or later. Typically, skeletal dysplasia and growth retardation are recognized in the first year or two of life; other manifestations include episodes of liver failure, renal dysfunction, and evidence of pancreatic exocrine insufficiency, neutropenia with recurrent infections, hypothyroidism, and mental retardation. The basis for this condition are mutations in the EIF2AK3 ( e ukaryotic translation i nitiation f actor 2 a lpha – k inase 3) gene (also known as PKR-like endoplasmic reticulum kinase or PERK ), which participates in the unfolded protein response in the ER. Environmental factors and modification by other genes may influence the spectrum of clinical severity. Skeletal dysplasia with bone fractures and episodes of liver failure plus NDM are pathognomonic of this entity; a review in 2010 indicated that fewer than 60 cases had been reported worldwide ; two novel mutations were subsequently reported. The prognosis is poor for affected patients. Treatment of the diabetes mellitus by pump therapy is recommended; parents should have genetic counseling for risks of recurrence, as this is an autosomal recessive condition.
FOXP3 : IPEX Syndrome
The term IPEX is an acronym for immune dysregulation, polyendocrinopathy, enteropathy, X-linked and is caused by dysfunction of T regulatory cells caused by mutations in FOXP3 , which normally is a transcriptional regulator for CD4 regulatory T cells, so that mutations result in the multiorgan autoimmunity. The clinical characteristics are early onset, insulin-dependent diabetes mellitus, or enteritis; eczema and elevated serum immunoglobin (Ig)E also occur early. Later manifestations include primary hypothyroidism, nephritis, hepatitis, enteritis, and alopecia. Bone marrow stem cell transplant offers potential for cure, but it depends on the availability of a suitable donor. Scurfy mice serve as a model for this condition in humans.
Further study of patients with NDM has revealed cases carrying FOXP3 mutations but without all features of the IPEX syndrome, as well cases with multiple autoimmune conditions in addition to diabetes attributable to mutations in other genes important for immune regulatory function: STAT1 , STAT3 , LRBA, and IL2RA .
GLIS : Neonatal Diabetes Mellitus With Congenital Hypothyroidism
GLIS3 is a member of the GLI-similar zinc finger protein family, which can function both as a repressor and activator of transcription and is specifically involved in the development of pancreatic beta cells, the thyroid, eye, liver, and kidney. An autosomal recessive syndrome, characterized by NDM, IUGR, congenital hypothyroidism, hepatic fibrosis with cholestasis, polycystic kidneys, and congenital glaucoma, was first described in 2003, and the responsible mutation in the GLIS3 gene was identified in 2006. As noted, this gene is expressed in the early developmental stages of the thyroid, eye, liver, kidney, and the pancreas, particularly in beta cells. Thus mutations in GLIS3 may have a role in other developmental anomalies and possibly in T1DM or T2DM. Novel mutations associated with the classical findings of NDM and hypothyroidism, plus bilateral sensorineural deafness, osteopenia, and pancreatic exocrine insufficiency, enlarge the clinical spectrum as reported. In animal studies, it appears that GLIS3 expression is required for pancreatic beta cell function and maintenance of beta cell mass in the adult, so that impaired function could lead to diabetes mellitus. Genome-wide association studies identify GLIS3 as a locus that affects risk for T1DM, and variants of GLIS3 have been implicated in the predisposition to T2DM.
PTF1A : Neonatal Diabetes Mellitus With Pancreatic and Cerebellar Agenesis
The gene PTF1A encodes pancreatic transcription factor 1 alpha known to be involved in pancreatic development. Mutations in this gene were identified in two families with NDM associated with pancreatic agenesis, as well as cerebellar agenesis, implicating this factor in normal cerebellar development, as well as pancreatic development, findings confirmed in knockout models of this gene in mice.
RFX6 : Mitchell-Riley Syndrome With Biliary and Intestinal Atresia
In mice, it was shown that absence of the gene encoding transcription factor Rfx6 prevents the formation of any pancreatic endocrine cells, except for pancreatic polypeptide producing cells; mutations in the human ortholog RFX6 gene caused an autosomal recessive form of neonatal diabetes mellitus. In humans, the syndrome of NDM, intestinal atresia, and hepatobiliary abnormalities had been known as the Mitchell-Riley syndrome . Clinically, patients are characterized by severe IUGR, fluctuations in glucose concentrations, stabilizing as persistent hyperglycemia and requiring insulin for treatment, cholestatic jaundice and anatomic findings of intestinal atresia, agenesis of the gall bladder, and abnormal formation of the pancreas. Quality of life may also be affected by mental retardation. Life expectancy for those who survive this critical neonatal period requiring intensive surgical and medical interventions is uncertain.
MNX1 and NKX2.2
A fascinating report identified mutations in two other transcription factors previously known to be important for pancreatic beta cell development: NKX2.2 homozygous mutations were found in two families with diabetes and severe developmental delay, hypotonia, cortical blindness, impaired visual tracking, and hearing impairment, whereas a more variable phenotype was found in two other families with homozygous MNX1 mutations where both patients had diabetes, but only one had additional features, such as severe neurological complications, hypoplastic lungs, and sacral agenesis.
NEUROG3 : Neonatal Diabetes Mellitus With Congenital Malabsorptive Diarrhea
Neurogenin 3 ( NEUROG3 ) is a critical transcription factor for the differentiation of islet endocrine cell types from the pancreatic endoderm. Mice lacking this factor are devoid of intestinal pancreatic endocrine cells and develop diabetes mellitus as newborns. In humans, homozygous mutations in the NEUROG3 gene cause malabsorptive diarrhea and early-onset diabetes mellitus.
GATA6 and GATA4
Both GATA4 and GATA6 are critical factors with differential contributions for normal pancreatic organogenesis; double Gata4 and Gata6 knockout mice do not develop a pancreas and hence have diabetes. In humans, heterozygous inactivating mutations in GATA6 were found to be the most common cause of pancreatic agenesis occurring in 15 of 27 subjects with pancreatic agenesis. These patients required both insulin plus pancreatic enzyme replacement. In a follow-up study, the same investigators sought GATA6 mutations in 171 subjects with NDM of unknown etiology, having already identified other mutations, including 15 with GATA6 mutations, in 624 subjects out of a cohort totaling 795. In the new cohort of 171 remaining subjects, an additional nine new cases of GATA6 mutations were identified for a total of 24 affected out of the original cohort of 795 subjects (3%). In these nine new cases, two had NDM but did not require pancreatic enzyme replacement, and one had TNDM. In addition, four parents were found to have GATA6 mutations, but the diabetes was diagnosed between the ages of 12 and 46 years. Several of the nine new subjects had subclinical pancreatic enzyme deficiency. Except for one of the parents, subjects with GATA6 mutations had extra pancreatic features, congenital heart malformations being found in 83%. Mutations in GATA4 were more recently shown to cause infancy-onset diabetes complicated by variable levels of pancreatic hypoplasia and exocrine insufficiency, pancreatic hypoplasia or complete agenesis, congenital heart defects, and neurodevelopmental delay. Thus GATA6 or GATA4 mutations can cause PNDM or occasionally TNDM or adult-onset diabetes, with variable exocrine pancreatic insufficiency from complete agenesis, subclinical, or no deficiency at all. Because the majority of cases manifest congenital heart defects, it is reasonable to consider genetic testing in any individual with a congenital heart defect and diabetes, regardless of the age of onset of diabetes. In evaluating a patient with NDM, pancreatic imaging can be useful in that a finding of pancreatic agenesis or hypoplasia will suggest the need for clinical evaluation of exocrine pancreatic insufficiency or other features that are associated with a more restricted list of genes that include GATA6 , GATA4 , PDX1 , and PTF1A , as well as EIF2AK3 , HNF1B , GLIS3 , NEUROG3 , and RFX6 .
Diagnosis and treatment of neonatal diabetes mellitus
NDM is extremely difficult to diagnose clinically, because the cardinal signs of diabetes resulting from osmotic diuresis—polyuria and polydipsia—are masked by the normal high-volume liquid diet of infancy, and clinicians may be reassured rather than worried when babies are feeding frequently and have lots of wet diapers. A diabetes diagnosis can be considered in neonates and infants who display IUGR in utero or at birth, or who fail to thrive despite apparent adequate intake with normal or copious urine output. A positive family history of diabetes mellitus (especially neonatal or infancy-onset) in one or both parents, or in prior siblings, points to a likely genetic cause; however, most cases will be caused by de novo mutations. The diagnosis is established by confirmation of sustained significant hyperglycemia and/or glycosuria, both readily available in hospitals or medical clinics. Recognition of diabetes mellitus within the first week of life often represents TNDM1, because of the differential expression of imprinted genes on chromosome 6q24, particularly if congenital defects, such as macroglossia and umbilical hernia, are present. Somewhat later recognition should also include consideration of defects in the K ATP channel, involving KCNJ11 and ABCC8 genes, as well as the INS gene; diabetes-associated autoantibodies need not necessarily be determined, unless the baby is close to or older than 6 months of age at the time of diabetes diagnosis, or if other signs of immune dysfunction are present.
Genetic testing is mandatory in any infant diagnosed with diabetes under 6 months of age and should be considered in patients diagnosed up to a year of age or with other features suggestive of an underlying monogenic cause. Although a majority of cases will be related to 6q24 or mutations in KCNJ11 , ABCC8 , and INS , consensus guidelines recommend urgent comprehensive testing of all known genes either with NGS panels or through a tiered approach. A cause can be found in approximately 80% or more of cases diagnosed under 6 months of age, as well as a less certain fraction of cases diagnosed after 6 months of age. A molecular diagnosis is critical not only for guiding the possibility of completely different medical management, but also for guiding monitoring and evaluation of other possible associated features, informing long-term prognosis and outcome, as well as family genetic counseling. Current methodology permits molecular diagnosis to be completed relatively quickly with initial results often available within 1 to 2 weeks. Although commercial testing is available in many countries, consultation with regional centers of expertise can help guide testing decisions and interpretation, as well as allowing for the accrual of accumulated knowledge about best treatment and outcome for these rare disorders. If needed, many of these centers also enable genetic testing without charge as a result of funding from various national agencies (see Available Resources section later).
Insulin therapy is the mainstay of initial treatment in all cases, with the goal of rapidly correcting metabolic derangement and establishing normoglycemia. Because patients will often have DKA and/or be managed in intensive care units, an IV insulin drip is a viable and effective initial choice, but because this route requires IV access, it is not ideal for the long term. Subsequently, or in the absence of DKA, subcutaneous insulin can be used with regimens similar to older patients, although it should be understood that neonates and infants are often extremely sensitive to very small doses of insulin. A multiple daily injection insulin regimen similar to any type 1 diabetes patient will be effective, but in most cases will require dilution of the typical U100 rapid-acting insulin, which is typically done as a 1:10 dilution (U10, or 10 units/mL). When the appropriate diluent provided by the manufacturer is used, the diluted insulin should be as stable as typical insulin. A very small dose (often the minimal feasible dose of 0.5 unit) of long-acting basal insulin is favored over giving a larger dose that may be tolerated while the infant is feeding frequently but may ultimately be more than appropriate for basal requirements and cause significant hypoglycemia as the baby sleeps better and starts to space out the timing of feedings. Another excellent option if available is an insulin pump, with which it is usually not necessary to use diluted insulin because of the extremely low doses of bolus and basal insulin that can be programmed.
Once the patient is stable and genetic testing has been sent, some experts recommend that a brief glyburide trial could be considered while the family receives diabetes education and preparations are being made for discharge. In the absence of consanguinity or features suggestive of a rare syndromic cause, the chances of carrying a sulfonylurea responsive mutation approaches 50%. A standard protocol ( Table 10.2 ) should be followed whereby the dose of glyburide is increased daily if hyperglycemia persists, and meanwhile correcting with appropriate modest doses of insulin to maintain reasonable glycemic control. It is important to discuss the relative risks and benefits with the family and be mindful of the high likelihood of failure. If no significant improvement of hyperglycemia is seen after a few days of increasing to a dose approaching 1 mg/kg/day, then the patient should resume insulin monotherapy pending genetic testing results.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


