The majority of monoclonal antibodies approved for clinical use display intrinsic antitumor effects that are mediated by one or more of the following mechanisms.
Cell-Mediated Cytotoxicity
As components of the immune system, effector cells such as natural killer (NK) cells and monocytes/macrophages represent natural lines of defense against oncologically transformed cells. These effector cells express Fcγ receptors (FcγR) on their cell surfaces, which interact with the Fc domain of IgG molecules. This family is comprised of three classes (type I, II, and III) that are further divided into subclasses (IIa/IIb and IIIa/IIIb).
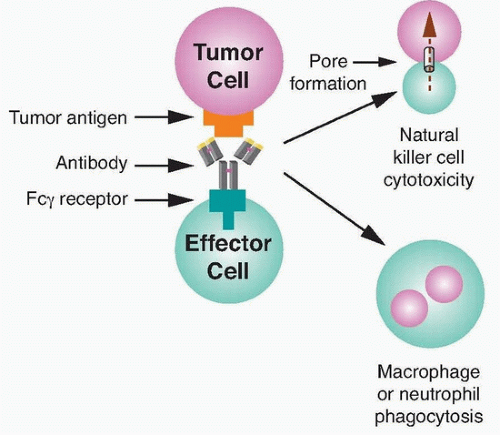
36 Recognition of transformed cells by immune effector cells leads to cell-mediated killing through processes such as ADCC and phagocytosis, as shown in
Figure 29.2, and can be mediated by FcγRI (CD64), a high affinity receptor capable of binding to monomeric IgG, or FcγRII (CD32) and FcγRIII (CD16), which are low affinity receptors that preferentially bind multimeric complexes of IgG. Signaling through type I, IIa, and IIIa receptors results in the activation of effector cells due to associated immunoreceptor tyrosine-based activation motifs (ITAM), whereas the engagement of type IIb receptors inhibits cell activation through associated immunoreceptor tyrosine-based inhibitory motifs (ITIM).
36 Clinical results support the idea that ADCC can play a role in the efficacy of antibody-based therapies. Naturally occurring polymorphisms in FcγRs alter their affinity for human IgG1 and have been linked to clinical response.
37,38 A polymorphism in the FCGR3A gene results in either a valine or phenylalanine at position 158 of FcγRIIIa. Human IgG1 binds more strongly to FcγRIIIa-158V than FcγRIIIa-158F, and likewise to NK cells from individuals that are either homozygous for 158F or heterozygous for this polymorphism.
39 The FcγRIIIa-158v was a predictor of early response and was associated with improved PFS.
A second polymorphism, FcγRIIa-131H/R, did not predict early response but was an independent predictor of time to progression (TTP).
38 Taken together, these data suggest that modulating the affinity of MAbs for FcγRIIIa, FcγRIIa, or both may increase the efficacy of therapeutic MAbs.
Each class of FcγR exhibits a characteristic specificity for IgG subclasses.
40 Many groups have focused on modifying the Fc domain of IgGs to optimize the engagement of subclasses of FcγR and the induction of ADCC, based on the findings of Shields et al.,
29 who performed a series of mutagenesis experiments to map the residues required for IgG1-FcγR interaction. Antibodies such as ocrelizumab, a humanized version of rituximab, have increased binding to low affinity FcγRIIIa variants and are now in clinical trials.
An alternative to modifying the Fc region of MAbs is to create bispecific antibodies (bsAbs) that recognize both a tumor-associated antigen and a
trigger antigen present on the surface of an immune effector cell.
43 Simultaneous engagement of both antigens can redirect the cytotoxic potential of the effector cell against the tumor.
41,42,43 Such antibodies are capable of eliciting effector function against tumor cell lines in vitro and in animal models. Two HER-2 directed bispecific antibodies, 2B1 and MDX-H210, have been tested in phase I clinical trials.
44,45Bispecific antibodies have a number of distinctive properties, including flexible choices of cytotoxic trigger molecules,
46 recruitment of effector function in the presence of excess IgG,
42 and custom tailoring of the affinity of the bsAb to match effector cell characteristics. These advantages have been facilitated by improved methods of bsAb production.
47 BiTE antibodies represent a novel class of bispecific, single-chain Fv antibodies.
48 Promising results have been seen in early phase clinical trials with at least two BiTE antibodies, one of which, blinatumomab, targets CD19/CD3.
49 Promising phase I results have also been reported in an interim analysis of an anti-EpCAM/anti-CD3 MT110 BiTE in the setting of advanced lung and gastrointestinal tumors.
50
Complement-Dependent Cytotoxicity
In addition to cell-mediated killing (see previous), MAbs can recruit the complement cascade to kill cells via CDC. Although IgM is the most effective isotype for complement activation, it is not widely used in clinical oncology. Similar to ADCC, the human IgG subclass used to construct a therapeutic MAb dictates its ability to elicit CDC; IgG1 is extremely efficient at fixing complement, in contrast to IgG2 and IgG4.
51 Antibodies activate complement through the classical pathway, by engaging multiple C1q to trigger activation of a cascade of serum proteases, which kill the antibody-bound cells.
52,53 The anti-CD20 MAb rituximab has been found to depend in part on CDC for its
in vivo efficacy.
54 Antibody engineering approaches have identified residues in the C
H2 domain of the Fc region that either suppress or enhance the ability of rituximab to bind C1q and activate CDC.
55 The ability to manipulate complement fixation through engineering approaches warrants
in vivo testing to determine the impact of these changes on the efficacy and toxicity of MAbs.