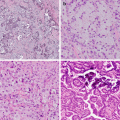
Fig. 15.1
Uncommon carcinomas. (a) Sarcomatoid carcinoma , pleomorphic carcinoma type with large cell undifferentiated carcinoma (left) and spindle cell pattern on the right. (b) Sarcomatoid carcinoma, spindle cell type. (c) Sarcomatoid carcinoma, pleomorphic carcinoma type, with squamous cell carcinoma (left) and giant cell carcinoma (center). (d) A pulmonary blastoma with fetal-type epithelial component (left), spindle cell component (right), and rhabdomyosarcomatous differentiation (inset). (e) Mucoepidermoid carcinoma with solid areas of bland intermediate cells and mucous cells with low-grade nuclei. Inset highlights mucicarmine-positive cells. (f) An adenosquamous carcinoma with adenocarcinoma with signet ring cells (left) and squamous cell carcinoma (right)
It has been proposed that this category of tumors represents a manifestation of epithelial-mesenchymal transition —that is, the potential of an epithelial cell to lose characteristic features of epithelium, becoming less cohesive and more like a mesenchymal cell. Evidence for common clonal origin of different components of these tumors and data favoring carcinomatous origins for these tumors will be discussed in the sections to follow.
Sarcomatoid Carcinoma (Except Pulmonary Blastoma)
As the name implies, these tumors have varied histology and include mixtures of carcinomatous components along with spindle or giant cell elements. While in the past this observation was of pathological classification and communication relevance alone, more recently, the recognition of the importance of adenocarcinoma (AdCa) and squamous cell carcinoma (SqCa) categories has revived interest in characterizing those components in PC. Mochizuki et al. [6] reviewed 68 cases of PC with AdCa component seen in 50%, SqCa in 16% and LCC in 34%. This distribution of AdCa component in PC was similarly reported by Nakajima et al. [7], with 49% containing AdCa, 22% SqCa and 16% LCC. Rossi et al. reviewed 75 sarcomatoid carcinomas with 51 PC showing 39% AdCa, 27% SqCa, and 41% LCC; their series had many cases with more than one pattern [8]. Overall, these data are similar to the original series of Fishback et al. [1]; among 61 cases of PC, 57% had AdCa, 10% had SqCa, and 32% had LCC.
IHC Characterization
The immunohistochemical characterization of PC suggests an epithelial origin with transition/progression to sarcomatous areas. In their analysis of 31 cases of PC, Pelosi et al. [9] showed cytokeratin, carcinoembryonic antigen (CEA), and epithelial membrane antigen (EMA) were more reactive in epithelial components and vimentin, fascin, and microvessel density greater in the sarcomatous component. However, immunoreactivity for cytokeratin and EMA were identified in pleomorphic components, and vimentin was seen in epithelial component. CEA was not positive in pleomorphic components. In a study extending IHC markers to lung-specific markers, Rossi et al. [8] showed spindle cell reactivity for thyroid transcription factor 1 (43%), cytokeratin 7 (62%), and surfactant protein A (6%) in PC, with similar rate of TTF1 (55%) and CK7 (70%) in spindle or giant cell carcinoma. In the same series, the epithelial component of PC was positive for CK7 (76%), TTF1 (59%), and surfactant protein A (39%); interestingly 39% of cases had AdCa histologically.
In a series of 19 cases of lung sarcomatoid carcinoma/PC [10], spindle or giant cell component was positive for keratin (80%), EMA (50%), p63 (50%), TTF1 (26%), and MOC31 (42%). Specifically in the ten cases of PC, the epithelial component was positive for keratin (100%), EMA (100%), TTF1 (70%), p63 (50%), and MOC31 (100%).
Histogenesis
Collectively, the IHC studies support the impression of epithelial/carcinomatous origin of PC. Existing data support a single clonal origin for sarcomatoid carcinoma, and therefore these tumors are carcinomas with sarcomatoid differentiation rather than sarcomas. While data on specific molecular events will be discussed in later sections, those relevant to histogenesis will be mentioned here.
In pleomorphic carcinoma, there is evidence that in the majority of cases, both epithelial and spindle cell component harbor the same KRAS mutation [11, 12] when a KRAS mutation is present. Individual cases have been reported in which a mutation was identified only in one component [13]. In carcinosarcomas , p53 mutational status was identical in both components [14].
Studies examining loss of heterozygosity at various loci support the contention that both elements in these biphasic tumors arise from the same clonal origin. The presence of more complex DNA changes in the mesenchymal component of PC and carcinosarcoma [15, 16] supports the view that the carcinoma component temporally precedes the mesenchymal component in the progression of the tumor.
Cytogenetics
Few comparative genomic hybridization studies have been reported in sarcomatoid carcinoma. Yakut et al. [17] performed CGH on AdCa , SqCa, LCC, and sarcomatoid carcinoma and showed that 5p gains were common to all groups, 3q gains seen in SqCa and in 2 of 4 sarcomatoid carcinomas , 14q gains in 3 of 4 sarcomatoid carcinomas which had overlap with AdCa, and 12p gains in LCC but not in other groups.
Specific Mutations in Sarcomatoid Carcinoma
TP53
Mutations in P53 were reported [14] in 4 of 9 spindle cell ca. (exon 5, 7, and 8) and 1 of 6 carcinosarcomas (exon 7). In a series of 22 PC, p53 mutation was seen in 14% of cases, most commonly exon 7 mutation [18]. Additional recent studies have shown a higher rate of TP53 mutations , from 58% [19] to 74% [20].
EGFR/KRAS Mutation
The identification of activating EGFR and KRAS mutations in lung AdCa and the association with response to EGFR-targeting tyrosine kinase inhibitors (TKI) have led to interest in the identification of these mutations in other histologic patterns of lung cancer. Given the admixture of patterns in pleomorphic carcinomas including cases with AdCa component and the proposed sequence of carcinoma to sarcomatoid pattern in these tumors, it would be expected that a proportion of PC would harbor these activating oncogenic mutations. Overall, in Asian populations in which EGFR mutations are seen at a higher rate than in Western populations, the rate of EGFR mutation is lower in PC when compared to AdCa. In 23 SC/PC, one European study [11, 21] identified two cases with EGFR mutation (9%); in 17 PC from Japan [13], three cases were identified (18%). Interestingly, all five of these cases had AdCa as the epithelial component. In a series of 22 cases of SC, Italiano et al. [21] reported no EGFR mutations; in that series, the histology of the epithelial component was not specified and the subtype of sarcomatoid carcinoma not reported. In one case report, an AdCa with exon 19 EGFR mutation was reported at autopsy as a pleomorphic CA with both the same activating exon 19 mutation and an acquired resistance T790 M mutation [22]. In two additional North American series of 33 and 36 cases, no EGFR mutations were identified [19, 23]. An EGFR mutation was described in one patient with carcinosarcoma, found in both adenocarcinoma and chondrosarcomatous elements [24].
In contrast, the rate of KRAS mutation in SC/PC has been similar to that of AdCa. In the aforementioned Japanese series of PC [13], no KRAS mutations were identified, similar to the relatively low rate reported in non-mucinous AdCa from Japan. KRAS mutation rates in Western series of SC have varied from 9 to 38%, with a combined overall rate of about 25% [11, 12, 19, 21–23, 25]. In the series of Pelosi et al. [12], the KRAS mutations were typical smoking-associated transversion mutations; of the mutation cases reported, four had an AdCa component, and two had LCC component. In a recent large study of 125 pulmonary sarcomatoid carcinoma, 34% harbored KRAS mutations, while EGFR mutations were seen in 5.6% (two were resistance-associated exon 20 insertions) [20]. One series suggested an adverse outcome among tumors with KRAS mutation [26].
Therefore, a subset of PC harbor mutations similar to AdCa , albeit with a relatively lower rate of EGFR mutation than seen in adenocarcinoma. This may be in part due to the smoking association of this tumor type. While EGFR mutation may suggest that PC/SC patients may benefit from EGFR-targeting TKI therapy, one caveat to this was reported by Shukuya et al. [27]. Response rate to TKI therapy among non-AdCa lung cancers harboring EGFR mutations was only 27% when compared to a 66% response rate in AdCa. While most of these cases were squamous, adenosquamous, and large cell histology, the three reported EGFR-mutated sarcomatoid carcinomas showed no response to EGFR TKI therapy. More recent studies support that response and durability of response in sarcomatoid carcinoma with EGFR mutations may be lower than in adenocarcinomas with these alterations [13, 22].
MET Exon 14 Skipping Mutations
Alterations in MET have been proposed as a precision medicine target for therapy with crizotinib . In 2009 [28], splice site and intronic mutations were identified in roughly 3% of lung AdCa that resulted in exclusion of exon 14 from the ultimate mRNA and resultant protein. These were exclusive of other driver alterations and are thought to activate MET by the loss of a critical Y1003 that is required for degradation of wild-type MET by CBL ubiquitin ligase. This loss results in resistance to degradation and persistence of MET activation. More recent studies confirmed this alteration in about 4% of lung adenocarcinomas [29–31].
A promising finding of activating MET exon 14 skipping mutations was reported in 8 of 36 sarcomatoid carcinomas (22%), with response to crizotinib in one patient [23]. This high rate in pulmonary sarcomatoid carcinoma was confirmed by two additional studies (27 and 32%, respectively) [32, 33], with a similar mutual exclusivity with other driver mutations. In a recent study of 125 pulmonary sarcomatoid carcinomas, 12% harbored MET exon 14 mutations, which was significantly enriched when compared to other carcinomas (3%). Of note these tumors more frequently contained an adenocarcinoma component as their epithelial component [20].
ALK Translocation and Amplification
While most series have not found ALK translocations in pulmonary sarcomatoid carcinoma [20, 23, 35], one study found three pleomorphic carcinomas and two carcinosarcomas with IHC reactivity for ALK (overall 3.5%) and FISH translocation, with one patient showing response to crizotinib . These patients were younger and had a lower level of cigarette smoke exposure [36]. In another series, one of 33 tumors (3%) harbored ALK translocation [19].
PDL1 Status
Sarcomatoid carcinoma has a higher rate of PDL1 immunoreactivity than other non-small cell lung carcinomas, with a rate over 50%. This, in addition to the presence of tumor-infiltrating lymphocytes [37], raises the possibility that these tumors will respond to immune checkpoint inhibitor therapies.
Sarcomatoid Carcinoma and Pulmonary Blastoma
The category of pulmonary blastoma (not pleuropulmonary blastoma, which is a pediatric tumor) will be separately discussed because of its relationship to well-differentiated fetal-type adenocarcinoma and, as a result, its unique molecular profile.
β-Catenin Mutations
The transcriptional effects of β-catenin promote decrease in cellular adhesion and increase in cellular migration. In nonneoplastic epithelial cells, β-catenin is part of a complex of proteins at the cell membrane; this complex includes E-cadherin , a critical component of intercellular adhesion. If β-catenin becomes disassociated from this complex, it is rapidly phosphorylated and targeted for degradation. With signaling via the Wnt pathway, this phosphorylation is inhibited, allowing accumulation of β-catenin and subsequent movement into the cell nucleus where it can exact its transcriptional effect. Mutations in β-catenin prevent its phosphorylation and therefore allow β-catenin accumulation and transcription effect in the absence of Wnt signaling.
For the diagnostic pathologist, non-mutated β-catenin localization by immunohistochemistry should be membranous; as a result of mutation, the nuclear immunoreactivity results from abnormal nuclear localization of β-catenin.
Nakatani et al. [38] described nuclear localization of β-catenin and β-catenin mutation in well-differentiated fetal adenocarcinoma and pulmonary blastoma. In a related study, Sekine et al. [39] demonstrated β-catenin mutations in 3 of 3 WDFA and 2 of 6 pulmonary blastomas. In a similar logic paralleling the identification of adenocarcinoma in PC and the association with KRAS/EGFR mutation, the presence of β-catenin mutations in WDFA and pulmonary blastoma suggests common histogenesis, arising from the carcinoma component.
Other Mutations
TP53 mutations were not identified in WDFA and were seen in one of seven PB studied (exon 6) [14]. In a series of five PB, three cases had β-catenin mutations, and no mutations were identified in KIT and KRAS. One case had both β-catenin mutation and an exon 19 EGFR mutation [40].
In a next-generation sequencing study of pulmonary blastoma, five patients showed mutations in BRCA2, BRAF, PTEN, EGFR, and PIK3CA. However β-catenin mutation or immunohistochemistry was not examined in this series [41].
In a study of three pulmonary blastomas (two of which harbored a β-catenin mutation), somatic DICER1 mutations were identified [42]. This is of interest as pleuropulmonary blastoma, the fetal/embryonic sarcomatous tumor of infancy, is associated with somatic and germline mutations in DICER1.
Carcinomas with Salivary Gland-Like Morphology
Less than 1% of lung c arcinomas represent tumors of bronchial gland origin that resemble their counterparts in the salivary gland. While they can occur in patients of any age, a large proportion occur in patients under the age of 30. As a result, they enter the differential diagnosis of carcinoid tumor, clinically and sometimes histologically. They are not thought to be smoking associated.
Mucoepidermoid Ca
The histology of mucoepidermoid carcinoma of the bronchus is similar to that of the salivary gland (Fig. 15.1e). Lower-grade tumors have cystic areas lined with mucinous cells with admixed intermediate cells; higher-grade tumors have less mucin, more intermediate cells, and atypical squamous cells resembling squamous cell carcinoma. In cases without clear-cut transition from low-grade mucoepidermoid carcinoma, the diagnosis of high-grade mucoepidermoid carcinoma shows considerable overlap with adenosquamous carcinoma.
Cytogenetics
The recognition of a recurrent translocation in mucoepidermoid carcinoma of the salivary gland (t(11:19)(q21;p13) involving chromosomes 11 and 19 [43] and the subsequent identification of a mucoepidermoid carcinoma translocated-1-mammalian mastermind like 2 fusion (MECT1-MAML2 fusion) [44] have introduced a relatively specific molecular marker for mucoepidermoid carcinoma. This has been subsequently examined in low- and high-grade tumors as well as tumors of different histologic subtypes. While lower-grade tumors harbor the translocation at a higher rate than the higher-grade tumors (75 vs 46%), other salivary gland and head and neck tumors do not demonstrate this translocation [36, 45]. As a result, there is speculation that some high-grade tumors that receive the diagnosis of MEC are in fact misclassified, and these misclassifications include adenosquamous carcinoma, squamous carcinoma, and salivary duct carcinoma.
Stenman [46] reported the same translocation in a child with pulmonary MEC, and this observation was confirmed in the lung [47] by the study of 17 pulmonary MEC by FISH and RT-PCR for MECT1-MAML2. An MAML2 rearrangement was confirmed in all low grade and 3 of 7 high-grade MEC by FISH (13 of 17 cases in all), but RT-PCR detected the fusion in only 6 of 14 cases. This difference in testing result may reflect variability in the fusion partner with MAML2. All cell types (mucous, intermediate, squamous) harbored the rearrangement. No cases of pulmonary AdSq (16), SqCa (24), or AdCa (41) had evidence of the translocation.
In a series of 18 pulmonary mucoepidermoid carcinomas, 12 of 18 were positive for MAML2 translocation, and this occurred in both low- and high-grade tumors [48]. Differences in survival were not seen based on MAML2 translocation status. In a detailed study of morphology, IHC, and cytogenetics, 24 mucoepidermoid carcinomas of the lung were associated with MAML2 translocation, and none of these had immunoreactivity for TTF1 or Napsin A [49]. This latter point is important in the distinction with adenosquamous carcinoma. In another study, MAML2 translocation was seen in 50% of the pulmonary MEC and tended to occur in younger patients, with tumors of low to intermediate grade [50].
While it appears that MEC is distinct from other lung tumors molecularly, the progression from low grade to high grade is not as clearly determined. Similar to the salivary gland, it raises the possibility that some of the high-grade MEC may be misclassified AdSq or SqCa.
EGFR/KRAS Mutation
Again, the attention regarding EGFR mutations in the treatment of adenocarcinomas of the lung has led to investigation of other tumors types. This has led to some interesting observations in MEC. In vitro data suggests a sensitivity to gefitinib in cells derived from MEC containing MAML2 translocation without EGFR mutation [51], and two reports of MEC tumors with gefitinib response in the absence of EGFR mutation [52, 53] have led to speculation that this tumor type may be responsive to EGFR TKI therapy. One confusing aspect of the latter study in the two reports is the discovery of L858R mutations in MEC in one series but no EGFR or KRAS mutation in another [53]. An additional study of 12 MEC showed chromosome 7 polysomy, EGFR immunoreactivity, but no amplifications or mutations in EGFR [54]. One question in these studies is again one of the misclassifications in high-grade tumors: without the identification of MECT1-MAML2 fusion, is the high-grade MEC actually AdCa or SqCa?
EML4-ALK Translocation
There is one report [55] of a low-grade MEC positive for EML4-ALK translocation . This report did not investigate MAML2 translocation.
Adenoid Cystic
The histology of adenoid cystic carcinoma of the lung is the same as its salivary gland counterpart, with cribriform structures, tubules, and nests. Some structures show “basement membrane” like material at the center of a nest of cells. These are also central tumors, without smoking association but with a slightly older average age at presentation than MEC.
There are few molecular studies of adenoid cystic carcinoma (ACC) of the lung. Because of frequent CD117 immunoreactivity in adenoid cystic carcinoma of salivary gland, Aubry et al. [56] studied pulmonary ACC with the finding that CD117 IHC is frequently seen. Mutations in KIT exons 9, 11, 13, and 17 were not identified in the12 cases studied. In a separate series of 12 adenoid cystic carcinomas, no EGFR amplification or mutation was identified [54]. In 24 cases, no mutations were seen in EGFR, KRAS, BRAF, PIK3CA, ALK, DDR2, or PDGFRA [57].
Alterations in chromosome 6 and chromosome 9 have been reported in adenoid cystic carcinoma and specifically in a bronchial ACC [58]. The finding of a MYB-NFIB fusion t(6:9)(q22–23;p23–24) in salivary gland type adenoid cystic carcinoma [59] has been reported and shown in pulmonary adenoid cystic carcinoma in 41% of cases; however, there does not appear to be a difference in morphology or outcome between translocation-positive or translocation-negative cases [60].
Adenosquamous Carcinoma
The definition of adenosquamous (AdSq) carcinoma requires presence of both adenocarcinoma and squamous cell carcinoma histology, with at least 10% of either component (Fig. 15.1f). Unlike MEC, these tumors show an association with cigarette smoking. This tumor type may become more frequent than previously reported (up to 4% of lung carcinoma) with the emphasis on histologic distinction of adenocarcinoma and squamous carcinoma with ancillary IHC. Also its distinction from high-grade MEC becomes relevant given the differences in molecular profiles and their impact on therapeutic decisions.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


