Fig. 5.1
An example of a PTT from Ref. [136]. Eight representative samples from de novo CRCs (T1 → T8), with truncating mutations identified by sequencing analysis. A substantial amount of normal full-length APC protein is seen from remaining normal alleles. The proteins were transcribed and translated in a reticulocytelysate system with 35S-methionine as a label. The kDa of the truncated proteins is predicted of the right
PTT has good sensitivity, a low-false-positive rate, and detects disease-inducing APC protein truncations, but not silent mutations, or the more than 20 APC gene polymorphisms, none of which cause disease [116, 136, 137]. PTT can also detect promoter mutations that lower APC mRNA expression and mutations, which create unstable mRNA (for those assays that amplify APC RNA). Disadvantages of the PTT are that it does not identify all APC gene mutations and does not identify the specific APC gene mutation, which is useful for directed mutation testing within effected kindreds. Last, PTT is superior to Western blotting, having a significantly higher APC protein detection rate [134]. Using this assay, Powell et al. [136] successfully identified APC mutations in 87 % 62 individuals with FAP tested. The PTT test is also used to identify mutations in other genes, such as BRCA1 and dystrophin [126, 138].
5.9.2 Direct APC Gene Sequencing and Multiplex Ligation-Dependent Probe Amplification
Direct DNA sequencing of the APC gene is considered the gold standard and detects APC gene mutations in ~70 % of individuals with FAP . The advantage of this technique is that all amplified bps are analyzed. However, the length of the APC coding region requires that many primers (~30) be used, making sequencing expensive, labor intensive, and time consuming [139]. Additionally, submicroscopic deletions have been detected in 6–55 % of APC kindreds, with larger deletions seen in 2 % and insertions seen in 6 % [115, 139, 140]. Since primer-dependent DNA sequencing is poor at detecting deletions/insertions, techniques such as multiplex ligation-dependent probe amplification (MLPA) have been employed to detect these APC mutations [140–142]. In MLPA, genomic DNA is interrogated with two probes that bind adjacent to each other. Each probe has one primer sequence that will amplify the two probes and intervening sequences only if the two probes are ligated. Once the two probes are hybridized to the genomic DNA, a ligation step is performed. If the two probes are hybridized and near enough to each other, the ligation step will produce one probe with an intervening sequence possibly revealing an insertion or deletion. Additionally, the probe will now have two primer sites allowing PCR amplification. The molecular weight of the amplified/ligated region is often measured via gel electrophoresis or by automated systems that measure a labeled and incorporated primer [140–144]. An example of MLPA is shown in Fig. 5.2.
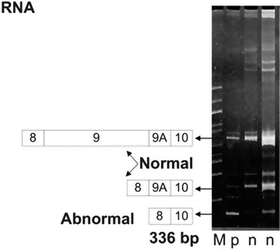
Fig. 5.2
An example of an MLPA analysis run on 6 % PAGE. M is a 100 bp ladder. The two n’s are normal splice variants of the APC gene; exons 8-9-9A-10 and 8-9A-10. p is from an individual with FAP showing a deletion resulting in a shorter 336 bp amplicon. The patient’s RNA was amplified by reverse transcription PCR and visualized with ethidium bromide staining
5.9.3 Conformation Strand Gel Electrophoresis and Single Strand Conformation Polymorphism Testing
Conformation strand gel electrophoresis and single strand conformation polymorphism testing (CSGE and SSCP, respectively) have both been used to detect APC gene mutations [105, 145]. In CSGE , control homoduplex wild-type dsDNA is compared to dsDNA, which may carry a point mutation. The two DNA duplexes are subjected to electrophoresis under mildly denaturing conditions. Under such conditions, heteroduplexes will often adopt a more complex three-dimensional conformation than homoduplexes and as a result migrate more slowly. In SSCP, wild-type and possibly mutant ssDNAs are compared. The two DNAs are denatured, rapidly renatured, and subjected to electrophoresis. A ssDNA segment with a point mutation will assume a different secondary and tertiary structure, resulting in a different migration pattern compared to the wild-type control. In general, CSGE and SSCP are less efficient and accurate than DNA sequencing; hence, sequencing has largely supplanted these testing methods. However, both still have value and have been used to screen for mutations that are then further analyzed via PTT or DNA sequencing [105]. Some data indicate that CSGE is more sensitive than SSCP [145].
5.10 FAP Management
FAP has an autosomal dominant inheritance pattern, and affected individuals have a virtually 100 % chance of developing CRC by their 1940s, making FAP detection and kindred analysis important in the management and prevention of FAP-associated CRC [105, 146, 147]. Once diagnosed, individuals with FAP should undergo annual lower endoscopy and have as many polys removed as possible. Upper endoscopy should be performed every 6 months to 3–4 years to detect duodenal, gastric, and periampullary adenomas, with the frequency determined by the polyp burden [146, 147]. Prophylactic colectomy is recommended by the end of the second decade, when there is usually a large polyp burden. A total protocolectomy with ileoanal J-pouch is recommended, although an ileorectal anastomosis can be performed on individuals with low rectal polyp numbers. With the latter procedure, yearly endoscopic rectal surveillance is still required [146–149]. Known individuals with FAP while under the age of 10 years should have annual α-fetoprotein levels and abdominal ultrasound to monitor for hepatoblastoma, 1 in 20 of which occur due to FAP [150].
Individuals with FAP given 400 mg/day celecoxib for 6 months showed a 28 % reduction in colorectal adenomas compared to a 4.5 % reduction in the control placebo group. Similarly, 400 mg celecoxib twice per day for 6 months reduced duodenal polyps 14.5 % compared to 1.4 % in placebo controls and 31 % in individuals with a high polyp burden [151, 152]. The US Food and Drug Administration Oncologic Drug Advisory Committee recommends celecoxib as an adjunctive treatment to endoscopic surveillance and colectomy in FAP treatment (New Drug Application Number 21,156).
Following a clinical FAP diagnosis, APC mutation testing should be performed. Standard APC mutation testing via DNA sequencing will identify mutations in 70–80 % of individuals clinically diagnosed with FAP [105]. MLPA and other methods will identify a further 2–5 % of mutations involving APC insertions/deletions, while MUTHY mutations will account for ~6.6 % of cases [105, 111, 141–144]. The remaining ~20 % of FAP have a more severe phenotype and appear to be due to APC mutations not identifiable by standard testing [111, 113]. Laken et al. [111] employed monoallelic mutation analysis to independently examine the status of each of the two APC alleles without mutations by standard testing. 78 % of the “unmutated” samples tested were found to have significantly reduced expression from one of their two APC alleles. Thus ~95 % of FAP is due to APC mutation. Monoallelic mutation analysis is complex and cumbersome, making it difficult to apply to molecular diagnostics.
Once a proband APC mutation is identified, all first degree relatives should have genetic counseling and molecular testing for the proband mutation. Individuals with FAP identified by an effected relative have a significantly greater life expectancy than individuals identified by FAP symptoms, warranting presymptomatic testing [153]. Directed mutation testing should be performed to simplify testing and lower costs [154]. Individuals with APC mutations should receive endoscopy, celecoxib treatment, and eventual colectomy. Individuals without mutations should receive endoscopic surveillance recommended for the general population.
When no APC mutation is detected in the proband, first degree relatives should receive annual lower endoscopic examines until age 35. If no colonic adenomatous polyps are found by age 35, lower endoscopic examines should be performed every 3 years until age 50, where endoscopic surveillance should match the general population. Where first degree relatives have a high burden of colonic adenomatous polyps, they should be managed as having FAP [105]. Presently, MUTYH, MLPA, or monoallelic mutation analyses are not routinely done for FAP testing [105, 111].
5.11 Attenuated FAP (AFAP)
AFAP is clinically defined by an autosomal dominant inheritance pattern, 10–99 adenomatous colonic polyps occurring at a later age than in FAP , and an 80–100 % lifetime CRC risk. Often, the colonic polyps are mostly proximal with little rectal involvement, increasing the likelihood of a misdiagnosis of sporadic polyps [105, 155]. Additionally, gastric and duodenal polyps occur and can rarely become malignant. Breast cancer and hepatoblastoma occur, with other extraintestinal manifestations being rare [155]. APC mutations are found in 20–30 % of individuals with AFAP and another 18 % have MUTHY mutations [105, 112]. APC mutations in AFAP are generally found 5′ or at the 3′ end of the gene, proximal to codon 1,517 or distal to codon 1,900 [105, 156–158]. Molecular testing for APC mutations in AFAP are done as for FAP. If testing reveals no APC mutation in individuals with AFAP, MUTHY testing should also be performed [105]. Due to the very high CRC incidence in AFAP, effected individuals and their first degree relatives should receive the same counseling and treatment as those with FAP [105].
5.12 Turcot and Gardner Syndromes
Turcot and Gardner syndromes are phenotypic variants of FAP . Gardner syndrome is characterized by colonic, gastric, and duodenal polyposis and soft tissue tumor, including osteomas, lipomas, fibromas, epidermal inclusion cysts, and desmoids tumors [159, 160]. APC mutations associated with Gardner syndrome are usually truncating mutations between codons 1,403 and 1,578 [160]. Molecular testing is the same as for other FAP variants [160].
Turcot syndrome is characterized by primary brain tumors accompanied by colorectal adenomas and/or CRC [161, 162]. Interestingly, Turcot syndrome shows molecular heterogeneity, with one form caused by APC mutations and the other resulting from HNPCC with mutations in one of the DNA mismatch repair genes MSH2, MLH1, PMS1, or PMS2 (see below, [162, 163]). In general, individuals with Turcot syndrome with APC mutations will have medulloblastomas, while those with HNPCC will have gliomas [162, 163]. Molecular testing for Turcot syndrome involves APC mutation analysis or DNA mismatch repair gene (MMR) analysis (see below).
5.13 I1307K APC Mutation in Ashkenazi Jews
Ashkenazi Jews carry a number of ancestral mutations conferring predisposition to several diseases, including CRC. One mutation found in 6–12 % of this population is the APC :I1307K mutation, involving a T → A transversion at nucleotide 3,920 converting AAATAAAA to an extended mononucleotide tract (A8), resulting in a lysine replacing isoleucine at codon 1,307 [164, 165]. While this amino acid substitution does not alter APC gene function, the A8 polymorphism is ~32 times more mutatable than the most mutatable wild-type APC sequences and often results in somatic biallelic APC inactivation and CRC tumorigenesis [164–166]. Interestingly, Gryfe et al. [166] found that, of the CRCs in APC:I1307K mutation carriers, only 42 % had APC-specific mutations that could be linked to the CRCs, a very modest mutation rate compared with HNPCC. Additionally, unlike in FAP , APC:I1307K must undergo somatic biallelic APC inactivation, resulting in only a 1.5–2-fold increased CRC incidence [164]. After adjustment for age, APC:I1307K carriers are more likely than noncarriers to have colorectal polyps, with either tubular adenomas or tubulovillous adenomas, but not villous adenomas, significantly more frequent among carriers (37.2 % vs. 23.6 %, P = 0.005) [166].
APC :I1307K mutation testing is limited to measuring a T → A transversion, making analysis via allele-specific oligonucleotide hybridization PCR a logical choice. For example, Niell et al. [167], employed a common reverse primer and two forward primers: one wild-type (5′-CTTTTCTTTTATTCTGC-3′) and one APC:I1307K (5′-CTTTTCTTTTTTTTCTGC-3′) specific primer. The 1 bp difference allowed the identification of the APC:I1307K transversion incidence in different populations. Other investigators have used PCR to amplify the genomic region around the APC:I1307K transversion and employed SSCP analysis, with transversion positive samples confirmed via DNA sequencing [168]. Not surprisingly, PCR has also been used to amplify the genomic region around the APC:I1307K transversion followed by direct amplicon sequencing [165].
Individuals with a positive APC :I1307K mutation test should undergo colonoscopic surveillance, as they may benefit from the early eradication of pre-cancerous polyps [169]. There is some evidence that smoking may increase CRC in APC:I1307K carriers, so smoking cessation should be encouraged [165]. The Ashkenazim population also have a low (0.7 %) carrier incidence of MSH21906G > C mutation, which is highly penetrant and causes CRC at an early age [164]. Last, although this population has a low (0.9 %) incidence of a BLM exon 10 frameshift mutation; BLM ASH . BLM ASH carriers do not have an elevated CRC incidence [165, 170].
5.14 MYH-Associated Polyposis
5.14.1 MUTYH and the MAP Phenotype
Reactive oxygen species derived from normal aerobic metabolism and exogenous sources readily damage DNA forming a wide variety of lesions, including oxidized bases [171–174]. One well-studied and common oxidized base is 7,8-dihydro-8-oxo-deoxyguanosine (8-oxo-G). 8-oxo-G readily pairs with adenine, leading to G:C → T:A transversion mutations following DNA replication [172–174]. MYH, the MUTYH gene product, is a glycosylase that catalyzes the cleavage of adenine mismatched to 8-oxo-G via base excision repair, preventing G:C → T:A transversions [175, 176]. Biallelic MUTYH mutation results in glycosylase activity loss and increased G:C → T:A transversions, including in APC and KRAS, but not Braf, leading adenoma development and CRC [177, 178].
MAP is the most important differential diagnosis for FAP and AFAP, occurring in ~1/10,000 individuals, accounting for ~0.6 % of all CRC [179, 180]. Phenotypically, biallelic MUTYH mutation (homozygous or compound heterozygous) results in a 93-fold increased CRC risk, with clinical symptoms appearing at 40–60 years, and a variable number of adenomatous colonic polyps, 10–100, rarely up to 1,000 [180]. Extraintestinal manifestations are less common than in FAP, with ~31 % of effected individuals having duodenal adenomas and/or gastric fundic polyps and a 4 % lifetime duodenal cancer risk [180–182]. MAP is autosomal recessive and therefore does not show the strong intergenerational history of FAP [180, 181]. The CRC incidence is high in MAP , with a 100 % lifetime risk and 50 % of effected individuals having CRC at diagnosis [181, 183]. Extraintestinal manifestations of MAP include breast, endometrial, and sebaceous carcinomas [182, 184]. Interesting, neither immunohistochemistry nor histology reveal any significant differences between MAP -associated CRC and sporadic CRC [185].
5.14.2 Molecular Testing for MAP
Unlike the 700+ known APC mutations, about 23 pathogenic MUTYH mutations are known, with two (Y165C and G382D), accounting for ~82 % of inactivating mutations [114, 115, 186]. Thus initial MAP testing is often directed at these two mutations. If one of these mutations is identified, sequencing is performed to identify the inactivating mutation on the opposite allele. If two inactivating mutations are found, the CRC risk will be very high. Failure to initially identify the Y165C or G382D combined with a strong suspicion for MUTYH mutation necessitates testing for less common mutations usually by DNA sequencing, which has the highest clinical sensitivity [187].
Numerous studies have demonstrated that in many cases ARMS is useful in MAP testing. For example, Piccioli et al. [188] employed a tetra-primer amplification refractory mutation system PCR (T-ARMS-PCR) to examine six common MUTYH mutations , which would identify at least 85 % of all mutations; Y165C, G382D, 1395_7delGGA, Y90X, 1103delC, and R231H. T-ARMS-PCR amplifies both wild-type and mutant alleles in one PCR reaction. T-ARMS-PCR and ARMS-PCR work well for MAP testing and cover ~85 % of known mutations [188, 189]. It is relatively inexpensive, can identity small deletions, requires no specialized equipment, and importantly is ideal for targeted molecular testing. Thus, once a kindred-specific mutation has been identified, ARMS-PCR and T-ARMS-PCR allow inexpensive, accurate, and rapid molecular analyses. Again ARMS-PCR does not identify ~15 % of known mutations, making DNA sequencing analysis necessary to identify uncommon mutations [187, 189]. The T-ARMS-PCR protocol is described in Fig. 5.3.
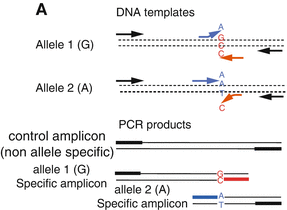
Fig. 5.3
T-ARMS-PCR from Ref. [188]. Two control primers (black arrows) which flank the mutation site, amplify one non-allele-specific control fragment, while two allele-specific primers in opposite orientations (blue and red arrows) in combination with the control primers, can amplify both smaller wild-type or mutant amplicons. The two allele-specific amplicons have different lengths and can easy be separated and identified via electrophoresis. Thus, the presents of wild-type and/or mutant amplicons can be determined with the longer non-allele-specific control amplicon serving as a positive control
5.14.3 MAP Management
For individuals with a significant colonic adenoma burden, MUTYH and APC mutation testing can be performed simultaneously, particularly if the family history suggests an autosomal recessive inheritance pattern. MUTYH testing should also be considered for the 20–30 % and 70–90 % of APC mutation negative FAP or AFAP cases, respectively. For FAP, the number due to MUTYH mutations is low, accounting for only ~5 % of FAP [111–114]. Once an individual is identified as having inactivating biallelic MUTYH mutations, molecular testing of all siblings is recommended as they have a 25 % chance of having the same mutation [112]. Presently, testing the partners of individuals with bilallelic MUTYH mutations is controversial, as the risk of MAP for children of an effected parent is ~1 % [181]. However if the partner of a bilallelic mutation carrier has one MUTYH mutation, an apparently autosomal dominant CRC inheritance is seen, possibly resulting in a misdiagnosis of FAP. Additionally, the CRC risk for MUTYH mutation heterozygotes is low, with a relative risk of 1.5–2.1 to the general population. Presently, the need for increased lower endoscopic surveillance is this population is undetermined [112, 190, 191].
Individuals with biallelic MUTYH mutations should undergo annual or biennial lower endoscopic surveillance beginning as teens. Upper endoscopic surveillance for duodenal adenomas/adenocarcinomas should be performed every 2 years [192]. The high rate of progression to CRC often necessitates a prophylactic colectomy [112, 192].
5.15 HNPCC
Lynch Syndrome , also known as HNPCC, is the most common hereditary CRC, accounting for ~2 % of all CRCs [193]. HNPCC was identified in 1996, when two large kindreds in the Midwestern United States showed excess CRC that lacked multiple colonic adenomas with a constellation of associated malignancies, particularly endometrial cancers [194]. Subsequent researchers named the disorder “Lynch Syndrome” or HNPCC. HNPCC is characterized by an autosomal dominant inheritance, high penetrance (~85 %), early onset CRC (~45 years), proximal CRC predilection, accelerated adenoma → CRC progression, increased survival with CRC, and CRC, which is histologically poorly differentiated, having increased mucinous and signet ring cell-features, and an excess of tumor-infiltrating lymphocytes. HNPCC associated extracolonic tumors include endometrial, ovarian, sebaceous, gastric, small intestinal, pancreatic, brain, hepatobillary tract, and upper urothelial cancers [193–195].
HNPCC results from activity loss of one or more DNA mismatch repair (MMR) genes, resulting in MSI, a specific form of genomic instability: [194, 195]. DNA polymerase activities collectively have a 10−7 error rate/bp synthesized. MMR gene activity locates and removes the substitution mismatches and insertion/deletion mismatches that arise during DNA replication. Additionally, the MMR genes play a role in dsDNA break repair (the most harmful DNA lesion), the initial sensing of DNA damage, and the initiation of apoptosis with severe DNA damage [171, 196, 197]. Overall, MMR gene activity increases genomic stability 50–103-fold, making MMR activity loss a powerful mutator phenotype [196]. The MMR genes are described below.
5.15.1 MutL Homolog 1 (MLH1)
MLH1 is located at 3p21.3-23 and consists of 19 exons of 57.36 kb, coding for an 84.6-kDa nuclear protein. MLH1 has two interaction domains, one which interacts with MSH2, MSH3, or MSH6 and the other which interacts with PMS2, MLH3, or PMS1. More than 250 different MLH1 germline mutations in NHPCC have been identified, consisting of missense, nonsense, splicing errors, and insertion/deletions, many of which truncated protein expression [198, 199]. Germline MLH1 mutations are seen in ~46 % of HNPCC cases, while 78 % of sporadic CRCs with high MSI (MSI-H) have reduced MLH1 expression secondary to promoter methylation and BRAF mutation. Thus MLH1 inactivation appears to occur by separate molecular pathways in HNPCC vs. sporadic CRC [200].
5.15.2 MutS Homolog 2 (MSH2)
MSH2 is located at 2p22-p21 and consists of 16 exons of 80.10 kb, coding for a 104.7 kDa nuclear protein. MSH2 has DNA, MSH3/MSH6, and MLH1/PMS2 binding domains. More than 300 different MLH2 germline mutations in HNPCC have been identified, consisting of mutation types similar to those seen in MHL1. MLH2 mutations are seen in ~40 % of HNPCC cases, thus ~90 % of HNPCC CRCs result from MHL1 or MLH2 mutations [198–202].
5.15.3 MutS Homolog 6 (MSH6)
MSH6 is located at 2p16 and consists of 10 exons of 23.8 kb, coding for a 160 kDa nuclear protein. MSH6 mutations account for 7–10 % of HNPCC [202–204]. 73 % of female MSH6 mutation carriers develop endometrial hyperplasia and carcinoma, compared to 29 % in MSH2 and 31 % in MLH1 carriers, making endometrial tumors more common among female MSH6 mutation carriers than CRC [205]. Most mutations are truncating mutations producing an inactive protein [205]. Interestingly, individuals with MSH6 mutations have a lower CRC incidence and a later age of onset than those with MSH2 and MLH1mutations [206].
5.15.4 Postmeiotic Segregation Increased 2 (PMS2)
PMS2 is located at 7p22.2 and consists of 15 exons of 63.14 kb, coding for a 96 kDa nuclear protein. PMS2 mutations account for less than 2 % of HNPCC kindreds and normally function as heterodimers with MLH1 forming MutLα [198, 202–204]. MSH2, MSH6, MLH1, and PMS2 associate with BRCA1, ATM, RAD50, MRE11, NBS1, and PCNA, forming a complex suggested to regulate DNA replication and maintain genomic integrity [207].
5.15.5 MutS Homolog 3 (MLH3)
Wu et al. [208] examined 39 unrelated HNPCC families who fulfilled the Amsterdam Criteria for HNPCC. Of 288 individuals examined, 12 had 10 different germline MLH3 variants, including one frameshift, and nine missense mutations. Normal immunostaining for MSH2, MLH1, or MSH6 was detected. The authors concluded that MLH3 mutation probably played a role in a small subset of HNPCC.
The molecular mechanism underlying CRC carcinogenesis in HNPCC appears to be increased genomic instability in ~32 target genes that have mononucleotide repeats of 7 or more within their coding sequences. Many of these genes, such as RAD50, BLM, and PTEN, are tumor suppressors; others such as TGFβ1R2 and BAX inhibit epithelial cell growth or exert proapoptotic effects. MSI causes frameshift mutations in these genes, leading to loss of function [209, 210]. Interestingly, MLH3, MSH3, and MSH6 have A9, A8, and C8 tracts, respectively, thus loss of one MMR gene may lead to progressive inactivation of the entire MMR system, possibly explaining the rapid adenoma → CRC progression seen in HNPCC [211].
5.16 HNPCC Diagnosis
Several guidelines have been developed to aid in HNPCC diagnosis: the Amsterdam criteria, the Amsterdam II criteria, and recently the Revised Bethesda Guidelines [212]. The Revised Bethesda Guidelines recommend MSI testing and/or immunohistochemical analysis in the following groups:
Colorectal cancer diagnosed in a patient less than 50.
Presence of synchronous, metachronous CRC, or other HNPCC associated tumors, regardless of age.
CRC with the MSI-H histology diagnosed in a patient less than 60.
CRC diagnosed in one or more first-degree relatives with an HNPCC-related tumor, with one of the cancers being diagnosed under age 50 years.
CRC diagnosed in two or more first-or second-degree relatives with HNPCC-related tumors, regardless of age.
Additionally, the following patients should also receive the same testing:
Patients diagnosed with Muir-Torre syndrome (association of CRC with cutaneous sebaceous neoplasms) or Turcot Syndrome (especially glioblastoma brain tumor).
Family members of individuals with a known MMR gene mutation.
5.16.1 HNPCC Testing Guidelines
Once HNPCC is suspected, specific testing protocols should be followed to obtain a definitive diagnosis. First, MSI or immunohistochemical analyses of CRC and normal tissue should be performed. Should MSI be present, or MMR protein expression lost in CRC, further analyses are performed to diagnose HNPCC [212, 213].
5.16.2 MSI Testing for HNPCC
MSI testing involves the analysis of five microsatellite markers standardized by the National Cancer Institute for HNPCC screening: BAT-25, BAT-26, D2S123, D5S346, and D17S250. Typically, both tumor and matching normal patient DNA are compared. MSI testing may be performed without normal patient DNA, but it is recommended that all studies assessing MSI using these markers should use matched normal-tumor DNA pairs [212]. Each microsatellite marker is PCR amplified with primers specific for each marker, and the resulting amplicons are analyzed for length variation via techniques such as gel electrophoresis, automated sizing equipment, or denaturing high-performance liquid chromatography. The primers may be labeled or unlabeled, depending on the requirements of the amplicon detection method [213–215].
MSI is defined as a change of any length due to a deletion/insertion of repeating units within a tumor microsatellite not seen in normal tissue DNA. With five markers, MSI is present and high (MSI-H) when ≥ 2 or more microsatellite markers show MSI. MSI is low (MSI-L) when one marker has MSI, and the tumor is MSI-L or microsatellite stable when one or no markers show MSI. Where more the five markers are analyzed, ≥ 30–40 % with MSI equals MSI-H, < 30–40 % is MSI-L, and no MSI is microsatellite stable [213]. Microsatellite stable or MSI-L tumors are unlikely to result from HNPCC [212]. An example of an MSI analysis is shown in Fig. 5.4.
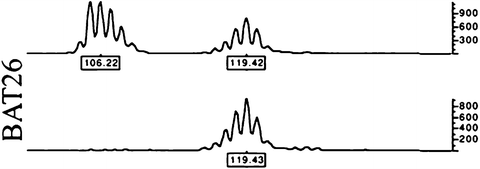
Fig. 5.4
An example of CRC MSI positivity at the BAT26 microsatellite from Ref. [212]. Genomic was PCR-amplified from tumor (upper) and normal (lower) tissue and run in an ABI PRISM 377 DNA Sequencer. The data were collected automatically and analyzed by GeneScan 3.1 software
5.16.3 Immunohistochemical Analysis for HNPCC
Immunohistochemical analysis for the loss of MMR genes is as effective as molecular MSI testing for HNPCC [212, 215, 216]. Immunohistochemical analysis of CRC and adjacent normal colorectal epithelium from individuals with HNPCC will often reveal loss of nuclear MSH2, MLH1, MSH6, or PMS2 protein expression. Following CRC analysis, if staining for each MMR protein is identified, the individual is unlikely to have HNPCC. If staining for an MMR protein is lost in CRC, but present in adjacent normal colonic epithelium, HNPCC is likely to be present [212, 215, 216]. The correlation between immunohistochemical MMR protein loss in CRC and MSI is high, with some studies showing MLH1 or MSH2 loss having 100 % specificity and 92.3 % sensitivity for predicting CRC with MSI-H [216]. Although both MSI and immunohistochemistry will work for initial testing, ideally both should be performed [212, 215]. Interestingly, both MSI-H and loss of MMR protein expression has been identified in a significant portion of colonic adenomas from individuals with HNPCC, demonstrating that these events occur early in CRC genesis [215]. Immunohistochemical testing has the advantage of being both cheaper and faster than MSI testing, with costs estimated to be 14–28 % of MSI testing [217]. There is growing interest in the application of reflex testing for MMR and/or MSI for all patients diagnosed with CRC. In particular, the Evaluation of Genomic application in Practice and Prevention (EGAPP) Working Group has provided genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch Syndrome in relative.
5.16.4 Molecular MMR and BRAF V600E Mutation Testing in HNPCC
With no loss of MMR protein immunopositivity, or MSI testing showing MSI-L/MMS, HNPCC is unlikely and no further testing is required [212]. Should MSI-H and/or MMR protein loss be identified, molecular testing should be performed for MMR gene mutations. Most protocols recommend MLH1 and MSH2 molecular testing, as these make up ~90 % of MMR gene HNPCC mutations [198–202, 212, 215, 216]. If immunohistochemistry detected loss of a specific MMR protein, molecular testing should be directed to the encoding gene [212, 215, 216].
Ten to fifteen percent of sporadic CRCs are MSI-H, have MLH1 protein loss, and have the specific BRAF-V600E mutation [82–86, 218, 219]. In these tumors, the MLH1 promoter is hypermethylated, leading to loss of protein expression, while the MLH1 sequence is unmutated [84, 85, 218, 219]. BRAF-V600E occurs only in sporadic CRCs and is not seen in HNPCC-associated CRCs [84, 85, 220, 221]. Therefore, MLH1 molecular testing is unnecessary where a BRAF-V600E mutation has been identified, unless there remains a high clinical suspicion for HNPCC [84, 85, 195, 220, 221]. Thus BRAF-V600E testing can rule out HNPCC, saving time and avoiding costly MMR gene molecular analysis [82, 84, 85, 220].
Molecular testing for BRAF-V600E is described above. Testing typically follows two approaches:
1.
With MSI testing, BRAF-V600E mutation testing should be performed. With no identified BRAF-V600E mutation, MLH1 and MLH2 mutation testing should be performed. If no MLH1 and MLH2 mutations are identified, MSH6 and PMS2 testing can be performed.
2.
Where immunohistochemistry for MMR protein expression is employed, the specifically lost MMR gene should be tested for mutations. If the lost MMR protein is MLH1, BRAF-V600E mutation testing should be done first and followed by MLH1 gene analysis if Braf is unmutated.
Failure to identify an MMR gene mutation usually rules out HNPCC, while identification of an MMR gene allows an HNPCC diagnosis. In some cases, a gene variation of unknown significance is identified. In other cases, no MMR gene mutation is identified, but the family history and/or individual’s presentation still strongly indicate HNPCC. Genetic testing will not identify all mutations leading to a HNPCC phenotype. Where an MMR gene mutation is identified, directed testing should be performed on the probands at-risk relatives and genetic counseling should be offered [195].
MMR gene analysis has been performed in a variety of ways, including Southern blotting, SSCP, DNA sequencing, MLPA, denaturing gradient gel-electrophoresis, quantitative PCR, and combinations of these methods [222–225]. Many germline MLH1 and MSH2 mutations are missense, nonsense, and frameshift mutations and are readily detected by these techniques. However, other mutations include large deletions and duplications [225–227]. While Southern analysis will detect large genomic rearrangements, it is also cumbersome, expensive, and time-consuming compared to other methods [222]. Charbonnier et al. [225] employed short fluorescent fragments to analyze MLH1 and MSH2 mutations, detecting deletions and duplications. 19 and 16 labeled primer sets were required for MLH1 and MSH2 analysis, respectively, with the reactions multiplexed and analyzed on a Gene scanner model 672 fluorescent fragment Analyzer. Control and patient electropherograms were superimposed and amplifications/deletions were detected by observing changes between superimposed control DNA and control and patient DNA. Most MLH1 and MSH2 mutations are unique with only a few common and recurring mutations identified, thus ARMS would be an extremely inefficient approach to MMR gene mutation analysis [226]. An example of the technique employed in Vasen et al. [228] is shown in Fig. 5.5.
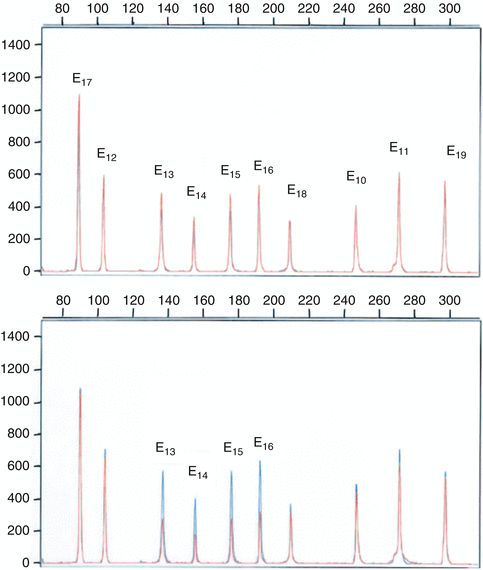
Fig. 5.5
The technique employed by Charbonnier et al. [226] to analyze exon deletions and duplications of the MMR genes in HNPCC CRC families, using multiplex polymerase chain reaction of short fluorescent fragments. Electropherograms from two individuals were superimposed. The Y axis displays fluorescence in arbitrary units, and the X axis indicates the size in bp. The upper diagram is a multiplex PCR of 10 MLH1 exons in two controls (in blue and red). The lower diagram shows detection of a genomic MLH1 deletion (in red) involving exons 13–16
5.16.5 HNPCC Management
Järvinen et al. [229] compared two at-risk HNPCC cohorts over a 15-year period. Colonoscopic screening reduced the CRC incidence 62 %. Additionally, the CRCs in the screened cohort were localized and caused no deaths, compared to 9 CRC-related deaths in individuals who declined colonoscopy. Interestingly, Vasen et al. [228] found several HNPCC patients with CRC 3.5 years following a negative colonoscopy, demonstrating a need for short screening intervals. Currently, individuals with HNPCC should receive colonoscopy every 1–2 years, beginning at age 20–25 (age 30 years with MSH6 mutation), or 10 years before the first effected family member presented with CRC.
In HNPCC, an increased incidence of CRC is found arising in flat and diminutive adenomas proximal to the splenic flexure. Interestingly, pan-colonic chromoscopic colonoscopy significantly increased flat and pedunculated adenoma detection in HNPCC. Presently however, pan-colonic chromoscopic colonoscopy is not routinely employed in the endoscopic surveillance of individuals with HNPCC, although it may allow a longer surveillance period prior to colectomy [230]. If endoscopic surveillance cannot be performed, a subtotal colectomy should be considered [231]. Prophylactic colectomy, whether subtotal or complete proctocolectomy, is not recommended if annual endoscopic surveillance can be carried out [231]. When unresectable adenomas or CRC are found, subtotal colectomy may be performed due the increased rate of secondary tumors [231].
Women with HNPCC have a 20–60 % lifetime risk for endometrial cancer and should undergo annual endometrial biopsy beginning at age 30–35 years. Transvaginal ultrasound has little value in screening for endometrial cancer, but can be used for annual ovarian screening [231–233]. Prophylactic hysterectomy/oophorectomy can be performed once the childbearing years are completed, as it results in a significant reduction in endometrial and ovarian cancer [234]. Screening for the more common Lynch Syndrome-associated malignancies, such as gastric cancer and urothelial cancer, is also recommended with frequency dictated by individual and family history (NCCN CRC screening guidelines). Screening for rarer malignancies is not defined by guidelines but is determined by individual and family history.
Most studies on NSAID use and chemoprevention in hereditary CRC have focused on FAP . There are no data on the effects of NSAIDS on chemoprevention in HNPCC, and they are currently not recommended. Additionally, although oral contraceptives are known to lower the incidence of endometrial and ovarian cancer in the general population, no data have demonstrated a chemopreventative effect in HNPCC. Last, individuals with HNPCC show an increased CRC with smoking and should be encouraged not to smoke [231].
5.17 Hamartomatous Polyposis Syndromes
Several heritable CRC syndromes are characterized by a specific constellation of extracolonic manifestations and increased hamartomatous rather than adenomatous polyps. Hamartomatous polyposis syndromes were first described in 1957 when investigators [232] reviewed the literature on pediatric on colorectal polyps and found that most contained mucous-filed glands with retention cysts, abundant connective tissue, and a high number of eosinophils. Presently, two hamartomatous polyposis syndromes with increased CRC are recognized: Juvenile polyposis syndrome (JPS ) and Peutz-Jeghers syndrome (PJS). Although both are diagnosed based on clinical manifestations, molecular tests are used to aid diagnosis.
5.17.1 JPS
JPS was first reported by Diamond et al. [235] who described a 30-month-old girl with a prolapsed polyp, accompanied by constipation, hematochezia, and a pedunculated sessile polyp on proctoscopy. In 1975, Stemper et al. [236] described a family with 10 affected members; juvenile polyps were identified in the stomach, small and large intestines, and rectum. Several developed gastric and CRC. Since that time, many studies have found increased gastrointestinal malignancies in kindreds affected by JPS , including gastric, colon, pancreatic, and small intestinal cancers [236–240]. The lifetime risk for malignancy in JPS is estimated to be 9–50 % [241, 242]. Histologically juvenile polyps are characterized by a smooth appearance, with cystically dilated glands embedded in a hyperplasic stroma which has an inflammatory infiltrate.
JPS is presently defined by any of three manifestations: (1) more than 5 juvenile polyps in the colorectum, (2) multiple juvenile polyps throughout the GI tract, and (3) any number of juvenile polyps and a family history of JPS [243]. JPS has been divided into three distinct categories: (1) JPS where the disease is limited to the colon, (2) JPS of infancy characterized by early onset, failure to thrive, hypoalbuminemia, and a poor prognosis, and (3) generalized JPS , where juvenile polyps are seen in the upper and lower gastrointestinal tract [244]. JPS is rare, occurring in ~1 in 105 individuals and shows an autosomal dominant inheritance pattern, with approximately 75 % of individuals having an affected parent and 25 % of cases arising de novo [245, 246]. The diagnosis of JPS is largely clinical, with molecular testing employed for prenatal diagnosis, kindred testing, and clinical diagnosis confirmation.
5.17.2 BMPR1A and SMAD4
SMAD4 or mothers against decapentaplegic homolog 4, is located at 18q21.1 and consists of 13 exons, which encode a 60.4-kDa protein of 552 residues. SMAD4 encodes an intracellular mediator of the TGF-β family and activin type 1 receptor signaling. Ligation of the TGF-β family receptor leads to SMAD1,−5 and−8 phosphorylation and activation, forming a complex with Smad4, activating pathways that mediate growth inhibitory signals [247–249]. Over 60 SMAD4 mutations have been identified in JPS . Interestingly, one mutation identified in multiple kindreds deletes 4 nucleotides (1244_1247delACAG), creating a truncated protein of 433 residues lacking regions critical for normal protein activity [249, 250].
BMPR1A or bone morphogenetic protein receptor, type IA, is located at 10q22.3, and consists of 13 exons that encode a 60.2 kDA protein of 532 residues. BMPR1A is a serine-threonine kinase, which upon binding TGF-β, phosphorylates and activates SMAD1,−5, and−8. These SMADs bind SMAD4 , creating a complex that inhibits cell growth and division [247–249]. Thus loss of either BMPR1A or SMAD4 activity results in cellular release from the TGF-β growth-inhibitory pathway, increasing malignant transformation risk [247, 248]. More than 60 BMPR1A have been identified, most of which produce a truncated, nonfunctional protein [247–250]. Together, BMPR1A or SMAD4 mutations account for ~45 % of all JPS cases [250]. Patients lacking BMPR1A or SMAD4 mutations are likely to have undiscovered gene mutations. Therefore, failure to identity BMPR1A or SMAD4 mutations does not exclude a JPS diagnosis. The identities of other genes mutated in JPS are currently unknown, although some data implicate endoglin (Eng) gene mutation [251].
5.17.3 Molecular Testing for JPS
Due to the large number of BMPR1A or SMAD4 mutations, DNA sequencing is most often employed in mutation analysis of these genes, although techniques such as PCR based denaturing gradient gel electrophoresis have been employed [249–251]. As for other genes analyzed, a set of specific forward and reverse primers first amplify the genomic DNA, which is purified, analyzed in a commercial sequencer, and compared to the wild-type sequence. Where a mutation is identified, it is often confirmed by sequencing in the opposite direction [249]. Such approaches have been able to identity 18 % and 21 % of BMPR1A and SMAD4 JPS mutations, respectively [249, 250]. As with most direct sequencing techniques, a number of deletions/insertions are missed; hence, techniques such as MLPA have been used to identify 6 % and 7 % more BMPR1A and SMAD4 JPS mutations, respectively [250]. Once identified, directed mutational testing can be performed on individuals related to the proband, who may be at risk for JPS .
5.17.4 JPS Management
Once an individual is identified as having JPS , a thorough history and physical examination should be performed, with particular emphasis on the present or absence of rectal bleeding/prolapse, anemia, constipation, obstruction, diarrhea, and abdominal discomfort. Symptomatic individuals should have upper and lower endoscopies, while asymptomatic individuals should have endoscopic screenings beginning at age 15, with all polyps removed. If the initial endoscopic examinations reveal no polyps, they should be repeated in 3 years. If polyps are found, endoscopies should be repeated every year, with all polyps removed. Once no more polyps are identified, endoscopic screening should performed once every 3 years. Individuals related to the proband without an identified mutation should been screened at age 15 and if no polyps are identified screening should be done 10 years to age 45 and then as the general population if no polyps are found [252].
Children with severe bleeding, iron-deficiency anemia, hypoproteinemia, failure to thrive, and/or a non-reducible intussusception should receive an ileorectal anastomosis [253]. Individuals without these severe symptoms should receive an ileorectal anastomosis by age 20–25, when the cancer risk becomes greater than risks associated with surgery. Individuals receiving an ileorectal anastomosis should undergo flexible sigmoidoscopy every 1–3 years for polyp formation in the rectal remnant [254]. Last, individuals with generalized JPS should undergo upper endoscopy every 3 years or annually if polyps are present, beginning at age 15, due to the increased upper gastrointestinal malignancies seen in generalized JPS [241, 242, 254]. Gastric polyps tend to be diffuse and difficult to endoscopically remove, hence a total of subtotal gastrectomy should be performed where gastric outlet obstruction, adenomatous changes, or adenocarcinoma are identified [254].
5.17.5 PJS
PJS is inherited in an autosomal dominant pattern occurring in ~1 in 60,000 and is characterized by pigmented mucocutaneous lesions, especially on the lips, buccal mucosa, and digits, as well as hamartomatous polyps mainly seen in the small intestine. The first detailed description of PJS was given by Peutz in 1921, who described a Dutch family with familial gastrointestinal polyposis, mucocutaneous lesions, and increased cancer [255]. Individuals with PJS have a ~15-fold increased risk for developing intestinal malignancies compared to the general population, with 50 % of PJS individuals dying of cancer by age 57 [255]. The increased malignancies include small intestinal, colonic, esophageal, gastric, breast, lung, pancreatic, uterine, ovarian, testicular, and cervical cancers. Other manifestations of PJS include intussusception, abdominal pain, intestinal polyp prolapse, and hematochezia [255–258]. Interestingly, women with PJS have a high incidence of cervical adenoma malignum and ovarian sex cord tumors with annular tubules, two very rare, well-differentiated neoplasms [255–258].
PJS polyps are characterized by bundles of smooth muscle between non-neoplastic epithelium, typically having an elongated arborizing, Christmas tree-like pattern. Endoscopically PJS polyps do not have specific features and their identification requires histological examination [255–258]. The diagnosis of PJS is based on clinical findings, although molecular-genetic testing is commonly performed for prenatal diagnosis, mutation-specific kindred testing, and clinical diagnosis confirmation. The syndrome is present when any of these conditions are met: (1) presenting with 2 or more PJS polyps, (2) having one polyp and typical PJS pigmented lesions, and (3) having one PJS polyp and a family history of PJS [259].
5.17.6 STK11/LKB1
PJS is caused by mutation of LKB1 or STK11 (serine/threonine kinase 11), found at 19p13.3. STK11 consists of 10 exons spanning 23 kb encoding a nuclear 48.6-kDa protein. STK11 is likely regulated by phosphorylation by cAMP-dependent protein kinase A and prenylation [260]. It functions as a tumor suppressor protein, with roles in the G1 checkpoint, p53-initiated apoptosis, and regulation of Wnt-β catenin-, vascular endothelial growth factor-, and mammalian target of rapamycin (mTOR) signaling pathways [261–265]. More than 140 different STK11 mutations have been identified in individuals with PJS, with many resulting in inactive truncated kinases. Germline STK11 mutations have been found in 50–70 % of PJS kindreds and 12–50 % of sporadic PJS cases [266–269]. The reason for the relatively low detection rate is likely due to genes other than STK11 being mutated in PJS and STK11 mutations being missed due to the limitations of specific molecular tests [270, 271].
5.17.7 Molecular Testing for PJS
STK11 mutation testing has been performed with a variety of techniques, including DNA sequencing (often limited to exons), CSGE, SSCP, Southern blotting, real-time PCR, quantitative fluorescent PCR, denaturing gradient gel electrophoresis, long range PCR, and MLPA [272]. Volikos et al. [273] analyzed STK11 mutations via MLPA and found 39 % (11 of 28 individuals with PJS) had large STK11 deletions ranging from loss of one exon to whole gene deletions. Taken together, ~59 % of STK11 mutations (range 10–100 %) are detectable by standard methods such as SSCP, CSGE, and DNA sequencing [269, 273–280]. Another 10 % of PJS cases are detectable by RNA based methods only and ~30 % of cases require MLPA testing to detect larger deletions [273, 274, 279, 280]. Thus, current molecular diagnostics allows 80–85 % STK11 mutation identification in PJS. Currently, most clinical testing employs sequencing of the 10 STK11 exons and/or MLPA, with the later technique most being effective at detecting larger deletions [272, 274].
5.17.8 Management of PJS
Due to their high risk for carcinomas, all individuals with PJS must be enrolled in lifelong cancer screening programs. While many individuals with PJS develop clinically apparent gastrointestinal lesions as early as their second decade, there are examples of malignancies in young patients and children [281, 282]. Presently there are no organization-approved guidelines for cancer surveillance in PJS patients and different institutions use different protocols [282]. The protocol used at the Johns Hopkins Hospital includes an initial baseline upper endoscopy and small bowel series at 8 years. If polyps are detected, upper endoscopy and small bowel series should be performed every 2–3 years. Upper endoscopy and small bowel series should be done annually from age 18. Colonic endoscopy should be performed every 2–3 years from age 18. At age 25–30, pancreatic endoscopic ultrasound should be done every 1–2 years. CT scanning and CA19-9 levels are offered as options. For women, a pelvic exam with a Pap smear should be performed annually from age 25. At this age, an annual transvaginal ultrasound and serum CA-125 should be done to screen for ovarian cancer. Last, at age 18, monthly breast exams should be done and at 25 semiannual clinical breast exams and annual mammography should be performed. Prophylactic mastectomy should be discussed with the patient. Males should have testicular exams and/or ultrasound of the testicles every 2 years until age 12 [283]. Recently, double balloon enteroscopy and intra-operative enteroscopy have been used to both facilitate better removal of upper gastrointestinal polyps and improve small bowel surveillance [282].
The macrolide compound rapamycin is an effective inhibitor of mammalian target of rapamycin, a cell growth-promoting kinase hyperactivated in PJS [265]. Rapamycin decreases polyp formation in PJS lkb1 +/− mice [284]. Currently, there are several clinical trials examining the effect of rapamycin on polyp formation in PJS patients [282]. Similarly, PJS lkb1 +/− mice treated with the COX-2 inhibitor celecoxib show reduced new polyp formation and a decreased size of preexisting polyps [285]. Two of 6 PJS patients given 200 mg celecoxib twice daily for 6 months showed a significant reduction in gastric polyps, suggesting that COX-2 inhibition may have some value in treating PJS [285].
5.18 Chemotherapy Testing for CRC
Irinotecan is a standard chemotherapeutic agent for mCRC treatment, often used in the FOLFIRI regimen: infused 5-fluorouracil, leucovorin, and irinotecan [14, 15]. The effective irinotecan dose depends on the initial amount of drug infused and rate of drug elimination by uridine diphosphate glucuronosyltransferase [UGT1A1 , 286]. Patient response to irinotecan has been difficult to predict, and toxic reactions are relatively common [287]. For example, fatal events of up to 5.3 % during single-agents irinotecan treatment have been reported [288]. Recently, molecular diagnostics have been used to prospectively identify patients who may be at risk for toxic irinotecan reactions.
5.18.1 UGT1A1
Irinotecan is a topoisomerase 1 inhibitor approved by the FDA in 1994 [14, 15]. Irinotecan stabilizes the complex between topoisomerase 1 and DNA, causing DNA breaks, DNA synthesis inhibition, and apoptosis [289]. Irinotecan is itself relatively inactive, but is converted to the 100–1,000-fold more active metabolite 7-ethyl-10-hydroxy-camptothecin (SN-38) by tissue and serum carboxylesterase-converting enzymes [286, 290]. SN-38 is glucuronidated and inactivated by hepatic UGT1A1 [286]. The dose-limiting toxicities of irinotecan are severe diarrhea and myelosuppression [287, 288, 291]. The relative activity of UGT1A1 determines the effective in vivo concentration and exposure time to SN-38 [292]. Thus, decreased UGT1A1 expression may lead to a toxic reaction secondary to increased SN-38 levels and longer exposure time [287, 288, 291, 292].
UGT1A1 is located at 2p37, spans ~160 kb, and has at least 9 promoters and first exons that can be spliced to four common expressed exons, resulting in 9 different UGT1A1 enzymes, with the most common “wild-type” gene/protein designated UGT1A1*1 [293, 294]. More than 60 UGT1A1 coding and promoter variations have been identified, which lessen UGT1A1 enzyme activity [295]. The most common polymorphism, UGT1A1*28 , contains 7, rather than 6, TA promoter repeats resulting in 70 % lower UGT1A1 expression [296, 297]. TA7 homozygotes are relatively slow to glucuronidate/inactivate SN-38, TA6 homozygotes are more efficient, and TA7/TA6 heterozygotes show intermediate activity. As expected, individuals with TA7 homozygotes are more likely to have severe diarrhea and neutropenia following irinotecan therapy, compared to TA6/TA6 and TA7/TA6 individuals [298]. Interestingly, individuals homozygous for TA5 and TA8 promoter repeats occur in African populations, with TA8 associated with Gilbert syndrome and lower in vitro glucuronidation rates [299].
Other polymorphisms, such as variants at codons 71, 486, 229, and 233, have amino acid substitutions that significantly reduce enzymatic activity and SN-38 clearance. For example, a T → G transversion at codon 486 (UGT1A1 *7) substitutes a tyrosine moiety for an aspartic acid, resulting in 93 % reduce enzyme activity [300, 301]. Following irinotecan therapy, UGT1A1*28 homozygotes show an increased incidence of irinotecan toxicity compared to UGT1A1*1 homozygotes, with other variants such as G71R (UGT1A1*6) predictive of severe irinotecan-induced neutropenia and poor chemotherapeutic response in Asian populations [302–304]. Many other polymorphisms exist that alter irinotecan therapy responses, often showing population-related distribution [303, 304]. Not surprisingly, individuals heterozygous for two low-activity UGT1A1 alleles often exhibit irinotecan toxicity. For example, Takane et al. [305] reported severe irinotecan toxicity in a UGT1A1*6/*28 patient.
5.18.2 UGT1A1 Molecular Testing
UGT1A1 allelic variations have been detected through a variety of techniques, including analysis of single base extension of PCR-amplified products, DNA sequencing, HRM, radioactive PCR, denaturing high performance liquid chromatography, and Invader technology. Currently, Invader technology is FDA-approved for UGT1A1 molecular testing and is commonly used [297, 302, 306–309].
Invader technology typically distinguishes between two different alleles within one non-PCR-based reaction. The reaction involves two steps. First, two allele-specific oligonucelotides, termed Invader oligonucleotides, bind to an unlabeled primary probe. The two Invader oligonucleotides hybridize in tandem with the primary probe forming a specific overlap structure. The 5′-end of the primary probe contains a non-complementary sequence that does not hybridize with either Invader oligonucleotide. The 3′-end of the Invader oligonucleotides are designed to overlap the primary probe such that one of the Invader oligonucleotides will form a complementary base pair with the primary probe. This bp (or pairs for the *28 TA7) correspond to the wild-type and polymorphism being tested. The complementary base pairing does occur, the resulting structure is recognized and cleaved by the Cleavace® enzyme (Flap endonuclease), releasing the 5′ portion of the primary probe. Should the 3′ portion of the Invader oligonucleotide not be complementary, the Cleavace® enzyme will not cut the structure.
In the second reaction, a second probe binds to the cleaved 5′primary probe sequence, forming a similar structure that is cleaved by the Cleavace® enzyme. The second probe is designed such that the second cleaving reaction separates a fluorophore from a quencher, creating an easily measured signal. This system can incorporate two primary probes that will eventually cleave either one of two secondary probes, each labeled with a spectrally distinct fluorophore corresponding to a specific allele. The ratio of one fluorophore to the other allows allele identification.
The accuracy of assay comes from the extreme specificity of the Cleavace® enzyme and the sequence specificity of the oligonucleotide hybridization. Purified genomic DNA may be used without PCR amplification and the system has the advantage of being completely closed once the assay has begun. Invader technology is a medium through-put, relatively low-cost technology that commonly allows up to 96 patient samples and controls to be simultaneously interrogated [308, 309].
Despite the large number of different techniques employed in UGT1A1 genotyping, the analytic sensitivity was 100 % for the UGT1A1*28 and 98–100 % for the *6 allele, where the referent test was DNA sequencing [310]. The reproducibility of Invader technology was 98.8 % [309]. Additionally, estimates of the first run failure UGT1A1*1 and UGT1A1*28 analysis was 5.0 % for sequencing, 1.7 % for PCR/capillary electrophoresis, and 6.7 % for Invader® technology, with most discrepancies solved by repeat testing [309].
5.18.3 UGT1A1 Polymorphism Management
The risk of a toxic irinotecan reaction is roughly 50, 12.5, and 0 % for *28/*28, *28/*1, and *1/*1 individuals, respectively [297]. However, there is significant overlap between these groups, and despite a large number of studies, the association between the *28 homozygotic genotype and irinotecan toxicity is not statistically significant [309]. Additionally, at standard irinotecan doses, the tumor response rate is 41 % for wild-type, 45 % for *28 heterozygotes, and 70 % for *28 heterozygotes [309]. Thus the wild-type individuals may be under dosed, while the *28 heterozygotes show a better response. Based on this, The Evaluation of Genomic Applications in Practice and Prevention Working Group found insufficient evidence to recommend for or against UGT1A1 genotyping guiding irinotecan treatment protocols [309].
Recently however, one Phase I study was performed on 27 patients with mCRC, examining irinotecan toxicity and the *28 polymorphism: 18 patients were *1/*1 and 9 were *1/*28. A maximum tolerated dose was not observed up to a 150 mg/m2 dose in the *1/*1 patients, while the *1/*28 patients showed toxicity at a 100 mg/m2 irinotecan dose [310]. The study concluded that *1/*1 individuals should receive 150 mg/m2 dose irinotecan, while *1/*28 individuals should receive 70 mg/m2. The study was limited to 27 patients and included no *28/*28 individuals. Most likely, further studies will be performed and may demonstrate clinical utility for UGT1A1 genotyping [310].
5.19 Future Directions
Molecular diagnostic testing in CRC is becoming increasingly important in the diagnosis and management of de novo and hereditary CRC, a trend that will probably continue [14–17, 60]. Several trends are readily apparent.
5.19.1 Increased Molecular Diagnostic Testing
Murff et al. [311] found that only 17 % of individuals who met the criteria for early-onset breast cancer testing were actually referred for genetic testing. Presently, many individuals who would benefit from molecular testing do not receive it. As the clinical utility of molecular testing becomes more apparent and testing costs fall, this diagnostic modality will be increasingly employed.
5.19.2 Expansion of the Number of Genes Tested in CRC
Presently, UGT1A1 genotyping to guide irinotecan use is not routinely performed, although an initial study suggests that it might have use in determining the optimal irinotecan dose [310]. Additionally, Bedoya et al. [312] found that Dukes stage C and D patients were significantly more likely (P = 0.05) to have a position 1,359 G → A nucleotide change in the cannabinoid receptor-1. They also had a shorter survival time and increased lymph node metastasis. While not currently a part of molecular diagnostics, increased understanding of mutations/polymorphisms in other genes effecting CRC genesis and progression, such as the cannabinoid receptor-1, will likely become important in the molecular diagnostics of this malignancy [14–17, 311].
5.19.3 Specific Molecular Techniques in CRC Testing and Analysis
Several molecular techniques, while not often employed in clinical testing, have proven to be very useful in the analyses of CRC and are very likely to significantly contribute to the molecular diagnostics of CRC in the near future.
Microarray. Micoarray consists of a solid support, often a glass slide, to which DNA probes of known sequence are fixed in an orderly arrangement. The slides are hybridized under high-stringency conditions with labeled nucleic acids (often cDNAs derived from RNAs), extensively washed, and the relative amount of probe-bound target sequence is measured via label detection. The number of gene/nucleic acid sequences that can be simultaneously measured can range from 10 to 1 million. Comparisons of normal colonic mucosa and CRC have shown a large number of gene products over-or under-expressed in CRC compared to normal colonic tissue [313]. Microarray has allowed the development of specific gene expression panels with diagnostic (Pathwork test for tissue of origin) or prognostic (OncotypeDX) abilities. For CRC, specific gene products have proven useful in predicting clinical behavior. For example, osteopontin RNA expression is a well-documented biomarker of CRC progression [314].
Proteomics . Proteomics involves the analysis and identification of proteins produced by cells in normal and pathologic conditions. Often these analyses examine protein modifications, such as phosphorylation and protein-protein interactions. The specific techniques used ranges from antibody-based protein chips resembling microarray techniques, to yeast two-hybrid systems to identify protein-protein interactions, high throughput crystallography screens for structural analyses, two-dimensional electrophoresis, and mass spectrometry. Although preliminary, these analyses have proven very useful in identifying proteins, whose expression correlates with chemotherapy response, metastatic potential, and degree of clinical aggressiveness. With time, specific proeomic profiles can be developed to predict the clinical behavior of specific CRC cases [313].
Related posts:
 Molecular-Genetic Testing in Penile, Scrotal, and Testicular Cancer
Molecular Diagnostics of Pancreatic Cancer
Molecular-Genetic Testing in Penile, Scrotal, and Testicular Cancer
Molecular Diagnostics of Pancreatic Cancer
 Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Pancreatic Endocrine Neoplasms
Molecular Pathology and Diagnostics of Cutaneous Malignancy
Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Pancreatic Endocrine Neoplasms
Molecular Pathology and Diagnostics of Cutaneous Malignancy
 Molecular Pathology and Diagnostics of Childhood Tumors
Molecular Pathology and Diagnostics of Childhood Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


