Molecular diagnostics is increasingly impacting a number of areas of cancer care delivery including diagnosis, prognosis, in predicting response to particular therapies, and in minimal residual disease monitoring. Each of these depends on detection or measurement of one or more disease-specific molecular biomarkers representing abnormalities in genetic or epigenetic pathways controlling cellular proliferation, differentiation, or cell death (Table 3.1). In addition, molecular diagnostics is beginning to play a role in predicting host metabolism of drugs—for example, in predicting fast versus slow thiopurine metabolizers using polymorphisms in the thiopurine methyltransferase (TPMT) allele and in use in dosing patients with thiopurine drugs.1 Molecular diagnostics has also had a major impact on assessing an engraftment after bone marrow transplantation and in tissue typing for bone marrow and solid organ transplantation.
The ideal cancer biomarker is only associated with the disease and not the normal state. The utility of the biomarker largely depends on what the clinical effect the biomarker predicts for, how large the effect is, and how strong the evidence is for the effect. For clinical application, biomarkers need a high level of analytic validity, clinical validity, and clinical utility. Analytic validity refers to the ability of the overall testing process to accurately detect and, in many cases, measure the biomarker. Clinical validity is the ability of a biomarker to predict a particular disease behavior or response to therapy. Clinical utility, arguably the most difficult to assess, addresses whether the information available from the biomarker is actually beneficial for patient care.
Biomarkers can take many forms including chromosomal translocations and other chromosomal rearrangements, gene amplification, copy number variation, point mutations, single nucleotide polymorphisms, changes in gene expression (including micro RNAs), and epigenetic alterations. Most biomarkers in widespread use represent either gain of function or loss of function alterations in key signaling pathways. Those that occur early and at a high frequency in tumors tend to be driver mutations, whose function is important for the cancer cell’s proliferation and/or survival. These are particularly useful as biomarkers because they often represent important therapeutic targets. However, cancer cells accumulate many genetic alterations, called passenger mutations, which tend to occur at a lower frequency overall and in a subset of a heterogeneous population of tumor cells that may contribute to the cancer phenotype but are not absolutely essential.2 Distinguishing passenger from driver mutations using various functional assays has become a major focus of translational research in cancer. The same biomarker may have utility in a variety of settings. For example, the detection of the BCR-ABL1 translocation, pathognomonic for chronic myelogenous leukemia (CML), is used for establishing the diagnosis, for the selection of therapy, and for monitoring for minimal residual disease during and after therapy.
Some of the most heavily used genetic biomarkers in cancer, particularly in hematologic malignancies, are chromosomaltranslocations. For certain diseases such as CML, detection of the BCR-ABL1 translocation or in Burkitt lymphoma the immunoglobulin gene-MYC translocation is required, according to current World Health Organization (WHO) guidelines, to make the diagnosis. Identification of translocations is important in the diagnosis and subtyping of acute leukemias (e.g., detection of PML-RARA and variant translocations in acute promyelocytic leukemia) and is also extremely important for the diagnosis of sarcomas such as Ewing sarcoma. The discovery of chromosomal translocations, such as the TMPRSS-ETS in prostate cancer and ALK translocations in non-small-cell lung cancer, portends an importance of detecting translocations in solid tumors.3 Chromosomal translocations, especially for hematologic malignancies, have been traditionally detected by classical karyotyping. This approach has limitations; in particular, it requires viable, dividing cells, which are often not readily available from solid tumor biopsies. In addition, a significant proportion of chromosomal translocations are not detectable by conventional karyotyping. For example, 5% to 10% of CML cases lack detectable t(9;22) by G banding. Such “cryptic” translocations require other approaches for detection, which are to be discussed, including fluorescent in situ hybridization (FISH), polymerase chain reaction (PCR), as well as nucleic acid sequencing-based methods.
In certain settings, it can be helpful to detect if a population of cells is clonal. For example, in some lymphoid infiltrates, the cells are well differentiated and it can be difficult to determine whether these represent a reactive or neoplastic infiltrate. If dispersed, cells are available and these could be analyzed by flow cytometer to detect whether a monotypic population expressing either immunoglobulin kappa or lambda light chains is present. In theory, immunohistochemical staining (IHC) for immunoglobulin light chains could be used to assess clonality; however, in practice this is done with more sensitivity using RNA in situ hybridization for immunoglobulin kappa and lambda light chain transcripts. The most sensitive way to detect clonality in a B-cell population is to analyze the size of the break point cluster region that arises as a result of VDJ recombination by PCR. Reactive B cells will show a distribution in the size of the VDJ recombination for the IGH or IGK or IGL, whereas clonal cells will show a predominant band that represents the size of the VDJ region of the dominant clone. Similarly, sometimes it can be difficult to distinguish neoplastic from reactive T-cell infiltrates. Given the large number of T-cell antigen receptors, it is not as simple to detect clonality by IHC or flow cytometry in T-cell proliferations. One approach is to use aberrant loss of T-cell antigen expression to aid in the diagnosis of T-cell neoplasms. Another is to detect clonal rearrangement of the VDJ region of the T-cell receptor gamma (TCRγ) gene, which can be done by PCR on both fresh and formalin-fixed paraffinembedded (FFPE) tissue.
Gene amplification is another important mechanism in cancer that has been found to have high utility in a subset of cancers. MYCN amplification occurs in approximately 40% of undifferentiated or poorly differentiated neuroblastoma subtypes,4,5 either appearing as double minute chromosomes or homogeneously staining regions. MYCN amplification is a very strong predictor of poor outcomes, particularly in patients with localized (stage 1 or stage 2) disease or in infants with stage 4S metastatic disease, where fewer than half of patients survive beyond 5 years.6
TABLE 3.1 Genomic Alterations as Putative Predictive Biomarkers for Cancer Therapy
Genes
Pathways
Aberration Type
Disease Examples
Putative or Proven Drugs
PIK3CA,51,52PIK3R1,53PIK3R2, AKT1, AKT2, and AKT354,55
Use of other chromosome abnormalities has been largely limited to the diagnosis and prognostication of hematologic disorders. Roughly half of all myelodysplastic disorders show cytogenetically detectable chromosomal abnormalities, such as monosomy 5 or 7, partial chromosomal loss (5q-, 7q-), or complex chromosomal abnormalities. Certain abnormalities in isolation (e.g., 5q-) have a favorable prognosis, whereas many others (e.g., “complex” karyotypes with three or more abnormalities) carry a worse prognosis. Differences in ploidy have proven to be useful predictors in pediatric acute lymphocytic leukemia (ALL), with hyperdiploid cases (>50 chromosomes) showing a distinctly more favorable course compared with hypodiploid or near diploid cases.7 Overall, DNA ploidy can be assessed by flow cytometry. Specific chromosomal copy number alterations can be detected by conventional karyotyping, array hybridization methods, or FISH.
Copy number variation (CNV) represents the most common type of structural chromosomal alteration. Regions affected by CNVs range from approximately 1 kilobase to several megabases that are either amplified or deleted. It is estimated that about 0.4% of the genomes of healthy individuals differ in copy number.8 CNVs resulting in deletion of genes such as BRCA1, BRCA2, APC, mismatch repair genes, and TP53 have been implicated in a wide range of highly penetrant cancers.9,10 CNVs can be detected by a variety of means including FISH, comparative or array genomic hybridization, or virtual karyotyping using single nucleotide polymorphism (SNP) arrays. Increasingly, CNV is detected using next-generation sequencing.
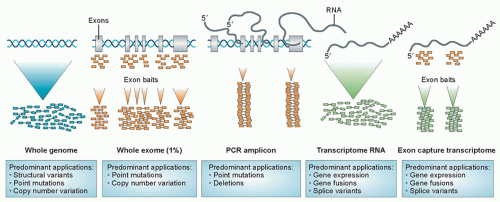
Large-scale sequencing of tumors has identified many mutations that are of potential prognostic and therapeutic significance. As will be discussed further, a wide range of strategies is available for the detection of point mutations (Fig. 3.1). It is important to recognize that many nucleotide variations occur at any given allele in populations. Formally, the term polymorphism is used to describe genetic differences present in ≥1% of the human population, whereas mutation describes less frequent differences. However, in practice, polymorphism is often used to describe a nonpathogenic genetic change, and mutation a deleterious change, regardless of their frequencies.
Mutations can be classified according to their effect in the structure of a gene. The most common of these disease-associated alterations are single nucleotide substitutions (point mutations); however, many deletions, insertions, gene rearrangements, gene amplification, and copy number variations have been identified that have clinical significance. Point mutations may affect promoters, splicing sites, or coding regions. Coding region mutations can be classified into three kinds, depending on the impact on the codon: missense mutation, a nucleotide change leads to the substitution of an amino acid to another; nonsense mutation, a nucleotide substitution causes premature termination of codons with protein truncation; and silent mutation, a nucleotide change does not change the coded amino acid.
Loss of function mutations, either through point mutations or deletions in tumor suppression genes such as APC and TP53, are the most common mutations in cancers. Tumor suppression genes require two-hit (biallelic) mutations that inactivate both copies of the gene in order to allow tumorigenesis to occur. The first hit is usually an inherited or somatic point mutation, and the second hit is assumed to be an acquired deletion mutation that deletes the second copy of the tumor suppression gene. Promoter methylation of tumor suppressor genes is an alternative route to tumorigenesis that, to date, has not been commonly employed for molecular diagnostics.
Oncogenes originate from the deregulation of genes that normally encode for proteins associated with cell growth, differentiation, apoptosis, and signal transduction (proto-oncogenes, [e.g., BRAF and KRAS]). Proto-oncogenes generally require only one gain of function or activating mutation to become oncogenic. Common mutation types that result in proto-oncogene activation include point mutations, gene amplifications, and chromosomal translocations. One example is mutations in the epidermal growth factor receptor (EGFR) that occur in lung cancer, which are almost exclusively seen in nonmucinous bronchoalveolar carcinomas. Somatic mutations of EGFR constitutively activate the receptor tyrosine kinase (TK). Importantly, responsiveness of tumors harboring these mutations to the inhibitor gefitinib is highly coordinated with a mutation of the EGFR TK domain.11,12
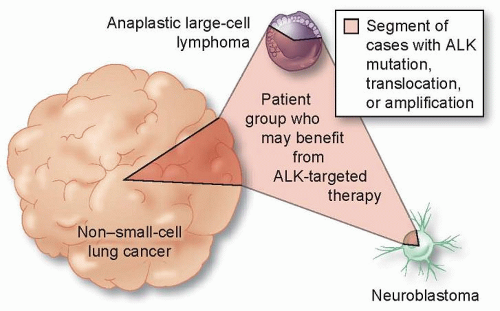
One of the challenges with using mutations as biomarkers is that there can be many nucleotide alterations that affect a given gene. For example, there are over 100 known different point mutations in EGFR reported in non-small-cell lung cancer. Many of these mutations occur at low frequency and have an unknown clinical significance.13,14 Another important concept is that the same driver oncogene may be mutated in a variety of different tumors. For example, lung cancers harbor a number of other different alterations that are common in other solid tumors, which generally occur at lower frequencies than EGFR mutations such as KRAS, BRAF, and HER2. Some lung cancers have translocations involving the ALK kinase gene. ALK, interestingly, is also activated by point mutations in a neuroblastoma as by translocation in anaplastic large cell lymphoma (Fig. 3.2). Hence, a therapy targeted to a genetic alteration in one cancer may demonstrate efficacy in other cancers.
The detection of mutations is also important in the evaluation of chemotherapy resistance. Roughly a third of CML patients are resistant to the frontline ABL1 kinase inhibitor imatinib, either at the time of initial treatment or, more commonly, secondarily. In cases of primary failure or secondary failure, over 100 different ABL1 mutations have been identified, including particularly common ones such as T315I and P loop mutations. While some mutations, such as Y253H, respond to second generation TK inhibitors (TKI), others, such as the T315I mutation, are noteworthy because they confer resistance not only to imatinib, but also to nilotinib and dasatinib.
Figure 3.2 Activating genomic alterations occur in a variety of tumor types. ALK translocations, mutations, and amplifications occur in non-small-cell lung cancer, neuroblastomas, and in anaplastic large cell lymphomas. Such recurrent alterations in cancer, together with effective inhibitors of these pathways, are transforming oncologic therapies from organ-specific to pathway-specific interventions and are driving the use of molecular diagnostics in a wider range of tumor types. (Modified from McDermott, U. and Settleman, J. J Clin Oncol 2009;27:5650-5659.)
Mutations are also used as important predictive biomarkers (Table 3.1). Two of the most notable examples are the use of the BRCA1 and BRCA2 mutation analysis for women with a strong family history of breast cancer. Over 200 mutations (loss of function point mutations, small deletions, or insertions) occur in BRCA genes, which are distributed across the genes necessitating full sequencing for their detection. The overall prevalence of these occur in about 0.1% of the general population.15,16 The lifetime risk of breast cancer for women carrying BRCA1 mutations is in the range of 47% to 66%, whereas for BRCA2 mutations, it is in the range of 40% to 57%.17,18 In addition, the risk of other tumors including ovarian, fallopian, and pancreatic cancer is also increased. Detection of BRCA1 and BRCA2 mutations is, therefore, important for cancer prevention and risk reduction.
THE CLINICAL MOLECULAR DIAGNOSTICS LABORATORY: RULES AND REGULATIONS
Laboratories in the United States that perform molecular diagnostic testing are categorized as high-complexity laboratories under the Clinical Laboratory Improvement Amendments of 1988 (CLIA).19 The CLIA program sets the minimum administrative and technical standards that must be met in order to ensure quality laboratory testing. Most laboratories in the United States that perform clinical testing in humans are regulated under CLIA. CLIA-certified laboratories must be accredited by professional organizations such as the Joint Commission, the College of American Pathologists, or another agency officially approved by the Centers for Medicare & Medicaid Services (CMS), and must comply with CLIA standards and guidelines for quality assurance. Although the regulation of laboratory services is in the U.S. Food and Drug Administration’s (FDA) jurisdiction, the FDA has historically exercised enforcement discretion. Therefore, FDA approval is not currently required for clinical implementation of molecular tests as long as other regulations are met.20,21
SPECIMEN REQUIREMENTS FOR MOLECULAR DIAGNOSTICS
Samples typically received for molecular oncology testing include blood, bone marrow aspirates and biopsies, fluids, organ-specific fresh tissues in saline or tissue culture media such as Roswell Park Memorial Institute (RPMI), FFPE tissues, and cytology cell blocks. Molecular tests can be ordered electronically or through written requisition forms, but never through verbal requests only. All samples submitted for molecular testing need to be appropriately identified. Sample type, quantity, and specimen handling and transport requirements should conform to the laboratory’s stated requirements in order to ensure valid test results.
Blood and bone marrow samples should be drawn into anticoagulated tubes. The preferred anticoagulant for most molecular assays is ethylenediaminetetraacetic acid (EDTA; lavender). Other acceptable collection tubes include ACD (yellow) solutions A and B. Heparinized tubes are not preferred for most molecular tests because heparin inhibits the polymerase enzyme utilized in PCR, which may lead to assay failure. Blood and bone marrow samples can be transported at ambient temperature. Blood samples should never be frozen prior to separation of cellular elements because this causes hemolysis, which interferes with DNA amplification. Fluids should be transported on ice. Tissues should be frozen (preferred method) as soon as possible and sent on dry ice to minimize degradation. Fresh tissues in RPMI should be sent on ice or cold packs. Cells should be kept frozen and sent on dry ice; DNA samples can be sent at ambient temperature or on ice.
For FFPE tissue blocks, typical collection and handling procedures include cutting 4 to 6 microtome sections of 10-micron thickness each on uncoated slides, air-drying unstained sections at room temperature, and staining one of the slides with hematoxylin and eosin (H&E). A board-certified pathologist reviews the H&E slides to ensure the tissue block contains a sufficient quantity of neoplastic tumor cells, and circles an area on the H&E slide that will be used as a template to guide macrodissection or microdissection of the adjacent, unstained slides. The pathologist also provides an estimate of the percentage of neoplastic cells in the area that will be tested, which should exceed the established limit of detection (LOD) of the assay.
MOLECULAR DIAGNOSTICS TESTING PROCESS
The workflow of a molecular test begins with receipt and accessioning of the specimen in the clinical molecular diagnostics laboratory followed by extraction of the nucleic acid (DNA or RNA), test setup, detection of analyte (e.g., PCR products), data analysis, and result reporting to the patient medical record (Fig. 3.3).
An extraction of intact, moderately high-quality DNA is essential for molecular assays. For DNA extraction, the preferred age for blood, bone marrow, and fluid samples is less than 5 days; for frozen or fixed tissue, it is indefinite; and for fresh tissue, it is overnight. Although there is no age limit for the use of a fixed and embedded tissue specimen for analysis, older specimens may yield a lower quantity and quality of DNA. Because RNA is significantly more labile than DNA, the preferred age for blood and bone marrow is less than 48 hours (from time of collection). Tissue samples intended for an RNA analysis should be promptly processed in fresh state, snap frozen, or preserved with RNA stabilizing agents for transport.
Dedicated areas, equipment, and materials are designated for various stages of DNA and RNA extraction procedures. DNA and RNA isolation can be done by manual or automated methods. Currently, most clinical laboratories employ commercial protocols based on liquid- or solid-phase extractions. Nucleated cells are isolated from biological samples prior to nucleic acid extraction. White blood cells (WBC) can be isolated from blood and bone marrow samples by different methods. One method involves lysing the red blood cells with an ammonium chloride solution, which yields the total WBC population and other nucleated cells present. Another method involves a gradient preparation with a Ficoll solution, which yields the mononuclear cell population only. Sections of FFPE tissue blocks are prepared for DNA extraction by first removing the paraffin and disrupting the cell membranes with proteinase K digestion. Fresh and frozen tissues also undergo proteinase K digestion prior to nucleic acid extraction. DNA isolation protocols consist of several steps, including cell lysis, DNA purification by salting out the proteins and other debris (nonorganic method), or by solvent extractions of the proteins with phenol and chloroform solutions (organic method). The DNA is then precipitated out of the solution with isopropanol or ethanol. The pellet is washed with 70% to 80% ethanol and then solubilized in buffer, such as Tris-EDTA solution. Proteinase K can be added to assist in the disruption and to prevent nonspecific degradation of the DNA. RNase is sometimes added to eliminate contaminating RNA. The DNA yield is quantitated spectrophotometrically, and the DNA sample integrity is visually checked, if necessary, on an agarose gel followed by ethidium bromide staining. Intact DNA appears as a high-molecular-weight single band, whereas degraded DNA is identified as a smear of variably sized fragments. After extraction, the DNA is stored at 4°C prior to use in a PCR assay, and is then stored at -70°C after completion of the assay. Because the DNA extracted from formalin-fixed tissue is degraded to a variable extent, an analysis of the extraction product by gel electrophoresis is not informative. Yield and integrity of the extracted DNA is best assessed by an amplification control to ensure that the quality and quantity of input DNA is adequate to yield a valid result.
Only gold members can continue reading. Log In or Register to continue