Molecular Mechanisms of Colorectal Carcinogenesis
Jinru Shia
Rhonda K. Yantiss
Colorectal carcinomas evolve via heterogeneous mechanisms and most arise from preexisting neoplasms, namely, adenomas. We now believe that colorectal cancer is essentially a genetic disease driven by an array of complex genetic and epigenetic perturbations.1 Cancer cells obtain early mutations that deleteriously affect genome stability and propagate additional changes, thereby generating a pool of alterations that enable cancer cells to proliferate and spread. Two major types of genomic instability have been widely studied in colorectal carcinoma: chromosomal instability and microsatellite instability (MSI). Elucidation of these mechanisms has been facilitated by study of two familial syndromes, familial adenomatous polyposis and Lynch syndrome (hereditary nonpolyposis colorectal cancer), respectively, resulting in recognition of several clinically important molecular aberrations. Some changes, such as mutations affecting tumor suppressor or DNA repair genes, aid identification of patients and family members with heritable cancer risk, whereas others have therapeutic implications, as discussed in Chapter 16.
KEY PLAYERS IN COLORECTAL CARCINOGENESIS
Oncogenes
Protooncogenes encode proteins that regulate embryogenesis and development, proliferation, differentiation, and apoptosis; and include growth factors, growth factor receptors, members of signal transduction pathways, and transcription factors. These proteins are normally produced in small quantities and rapidly degraded. Dysregulated protooncogenes are classified as oncogenes and develop via point mutations, chromosomal translocations, or other alterations. All of these changes increase gene expression, enhance protein activity, or confer resistance to degradation, resulting in an overall “gain of function” with respect to oncogenic activity. Thus, alteration of only one allele is sufficient to cause oncoprotein dysregulation.
Tumor Suppressor Genes
In contrast to oncogenes that drive cell proliferation and prolonged cell survival, tumor suppressor genes encode proteins that suppress transcription of protooncogenes and diminish the potency of their products. Tumor suppressor genes are highly conserved across species because their absence, or disrupted function, prevents inhibition of protooncogenic activity and leads to uncontrolled expansion of cell populations. Changes that affect tumor suppressor gene potency include mutations, translocations, loss of heterozygosity, and transcriptional silencing via promoter hypermethylation. In contrast to oncogenes that drive cell growth with only one altered allele, both tumor suppressor gene alleles must be defective in order to impact cell proliferation; the presence of one intact allele is generally sufficient to regulate protooncogenes. Most heritable cancer syndromes result from tumor suppressor gene germline mutations, in which case each cell harbors one dysfunctional allele. Loss or mutation of the second allele promotes neoplasia.
MicroRNAs
MicroRNAs comprise a recently recognized class of molecules that plays a major role in the posttranscriptional regulation of protein expression. These short, noncoding sequences participate in complexes that bind to, and prevent translation of, mRNA strands. Decreased expression of microRNAs that normally suppress translation of oncogenic mRNA transcripts will lead to an overall increase in oncoprotein activity. Alternatively, increased expression of microRNAs that regulate transcripts from tumor suppressor genes will further diminish regulation of cell growth. MicroRNAs are further discussed in Chapter 13.
APC/WNT SIGNALING AND THE CHROMOSOMAL INSTABILITY PATHWAY
Overview
Chromosomal instability underlies the stepwise genetic model developed to explain the progression from adenoma to invasive carcinoma.2 Biallelic inactivation of adenomatous polyposis coli (APC) on chromosome 5q facilitates adenoma formation, whereas subsequently mutations in KRAS, losses of large amounts of genetic material from chromosome 18q, and TP53 inactivation accompany the transition from adenoma to carcinoma (Figure 11.1). Tumors that develop via this mechanism are characterized by gains and losses of whole, or large portions of, chromosomes or loss of heterozygosity that results in karyotypic variability among tumor cells (i.e., chromosomal instability). The role of APC is of particular significance to this cancer model. Mutations in APC occur early in colorectal cancers and presumably exert their pathogenetic effects through a variety of mechanisms, including the Wnt signaling pathway as discussed below.
Familial Adenomatous Polyposis Due to APC Mutations
Familial adenomatous polyposis is a dominantly inherited condition. Patients with typical familial adenomatous polyposis have hundreds to thousands of adenomatous polyps in the colorectum, and virtually 100% of affected individuals develop colorectal cancer by the age of 30 years. Patients with
familial adenomatous polyposis are at risk for upper gastrointestinal polyps as well as a variety of extraintestinal manifestations. Two named syndromes represent subtypes of familial adenomatous polyposis with different extraintestinal manifestations: Gardner syndrome and Crail syndrome. Features of Gardner syndrome include osteomas, intraabdominal desmoid tumors (mesenteric fibromatosis), cutaneous cysts, lipomas, and dental abnormalities. The combination of polyposis and medulloblastomas is referred to as Crail syndrome, although this constellation of findings is sometimes mislabeled Turcot syndrome. Turcot syndrome is more appropriately used to describe a subtype of Lynch syndrome, in which patients develop microsatellite unstable intestinal cancers and glioblastoma multiforme, not medulloblastoma. Familial adenomatous polyposis patients are also at risk for hepatoblastoma, gastric adenocarcinoma, adrenal cortical adenoma, nasopharyngeal angiofibroma, and papillary thyroid carcinoma, particularly the cribriform-morular variant.3
familial adenomatous polyposis are at risk for upper gastrointestinal polyps as well as a variety of extraintestinal manifestations. Two named syndromes represent subtypes of familial adenomatous polyposis with different extraintestinal manifestations: Gardner syndrome and Crail syndrome. Features of Gardner syndrome include osteomas, intraabdominal desmoid tumors (mesenteric fibromatosis), cutaneous cysts, lipomas, and dental abnormalities. The combination of polyposis and medulloblastomas is referred to as Crail syndrome, although this constellation of findings is sometimes mislabeled Turcot syndrome. Turcot syndrome is more appropriately used to describe a subtype of Lynch syndrome, in which patients develop microsatellite unstable intestinal cancers and glioblastoma multiforme, not medulloblastoma. Familial adenomatous polyposis patients are also at risk for hepatoblastoma, gastric adenocarcinoma, adrenal cortical adenoma, nasopharyngeal angiofibroma, and papillary thyroid carcinoma, particularly the cribriform-morular variant.3
Most APC mutations are either nonsense or frameshift mutations.4 Nonsense mutations introduce a premature stop codon and produce a truncated protein, whereas frameshift mutations result from insertions or deletions. Large deletions affect bigger portions of the gene and result in loss of some, or all, of chromosome 5 (loss of heterozygosity). Different types of APC mutations are associated with variably severe gastrointestinal polyposis and extracolorectal manifestations. Approximately 70% to 90% of patients have truncating APC mutations that show a predilection for the mutation cluster region. The most severe phenotypes are associated with mutations in the c1290-1400 regions.
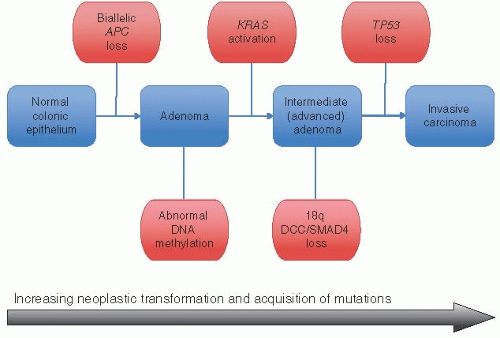 FIGURE 11.1: The chromosomal instability pathway of colorectal carcinogenesis proposes that adenomas develop as a result of biallelic inactivation of APC. Loss of APC function promotes additional genetic changes, including KRAS mutations, losses from chromosome 18q, and alterations affecting TP53. These subsequent changes typically occur via losses of large amounts of genetic material (loss of heterozygosity) and represent the molecular counterpart to the adenoma-carcinoma sequence. |
Deviation from the Classic Tumor Suppressor Model
The traditional “Vogelgram” model of colorectal carcinogenesis illustrates many principles that have proven true regarding colorectal neoplasia, but several developments since its description provide proof of variations to this paradigm. Attenuated forms of familial adenomatous polyposis are characterized by relatively few polyps (<100) and later onset of colorectal cancer. They result from mutations at the extreme 3’ (c78-167) and 5’ (c1581-2843) ends, producing a protein with partial function from the germline-mutated allele.5,6 A third hit mechanism is required for removal of residual APC function in these cases. The first hit is the germline mutation, the second hit inactivates the wild-type allele, and the third hit is a second, more traditional mutation in the germline-mutated allele that removes any residual function from the APC gene and promotes further tumor growth.6 A third hit also occurs in development of some sporadic colorectal carcinomas that contain one inactive and one partially functional allele for similar reasons.7
The concept that biallelic inactivation of APC occurs via independent events is also incorrect. Recent evidences suggest that the type of acquired APC mutation depends upon the location of the preceding germline alteration due to selection for at least one mutation between codons 1250 and 1450.8 Germline mutations in this region are followed by mutations in the second allele that result in complete loss of the gene (loss of heterozygosity), whereas germline mutations outside this region are associated with somatic mutations between codons 1250 and 1450, suggesting some level of interdependence between these events. Selection for at least one mutation between codons 1250 and 1450 ensures that mutant APC protein retains some of the 20 amino acid repeats required to bind β-catenin and provides adequate Wnt/β-catenin signaling to promote tumor formation.9 DCC apparently plays a less significant role in colorectal carcinogenesis than previously believed, as described below.
Familial Adenomatous Polyposis Due to MYH Mutations
Some patients with a colonic phenotype that suggests familial adenomatous polyposis do not have detectable APC mutations. Biallelic inactivating mutations in the base excision repair gene, MYH, have been identified as the cause of some previously unexplained polyposis cases. This gene is located at 1p32-34 and contains mutational hotspots, such that most germline mutations are Y165C and G382D missense mutations. Patients with inactive MYH acquire somatic APC mutations as a consequence of impaired base excision repair function. The MLH1 gene is also a reported target of MUTYH-associated transversions, in which case resultant tumors have the MSI phenotype.10
APC and Wnt Signaling in Colorectal Carcinogenesis
Activation of Wnt signaling occurs early in the progression of sporadic adenomas to adenocarcinomas of the colorectum.11 The APC protein product consists of 2,843 amino acids and performs several complex functions. It is normally expressed in nonproliferating colorectal epithelium and is essential for maintaining normal growth control and differentiation. Loss of APC function exerts a tumorigenic effect mainly through its impact on the Wnt signaling pathway, which is an evolutionarily conserved mechanism that interacts with several other signaling pathways important to colorectal neoplasia. In the absence of Wnt signaling, the APC-axin-GSK3β cytoplasmic complex induces phosphorylation of serine and tyrosine residues on β-catenin, thereby targeting it for ubiquitination and ultimate destruction and preventing its stimulating effects on cell proliferation. However, active Wnt signaling promotes the effects of β-catenin. Wnt proteins are likely secreted by adjacent subepithelial cells and bind to an epithelial cell surface receptor complex that includes the Frizzled receptor and the LDL receptor-related protein coreceptor.12 Coupling these molecules induces an interaction between axin and LDL receptor-related protein that drives phosphorylation of the cytoplasmic protein, Dishevelled. This protein subsequently interferes with the APC-axin-GSK3β cytoplasmic complex and prevents phosphorylation and ubiquitination of β-catenin, resulting in its cytoplasmic accumulation. Excess β-catenin is then translocated to the nucleus where it associates with T cell factor (TCF) and lymphoid enhancer factor, forming a complex that promotes expression of several important cell cycle regulating genes (Figure 11.2). Thus, APC negatively regulates Wnt signaling via
participation in the APC-axin-GSK3β destruction complex that promotes β-catenin degradation.13 Immunohistochemical stains for β-catenin can serve as surrogate markers of Wnt signaling. Staining for β-catenin is limited to epithelial cell membranes with weak cytoplasmic staining in the absence of Wnt signaling, but strong staining of the nucleus and cytoplasm suggests that the pathway is activated.
participation in the APC-axin-GSK3β destruction complex that promotes β-catenin degradation.13 Immunohistochemical stains for β-catenin can serve as surrogate markers of Wnt signaling. Staining for β-catenin is limited to epithelial cell membranes with weak cytoplasmic staining in the absence of Wnt signaling, but strong staining of the nucleus and cytoplasm suggests that the pathway is activated.
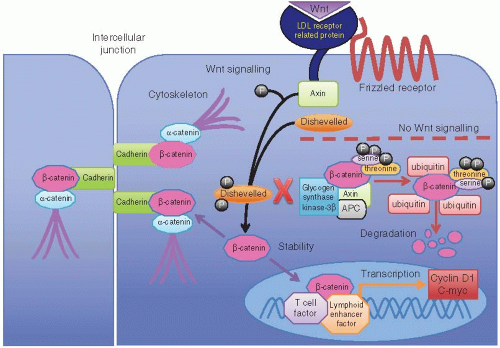 FIGURE 11.2: Cytoplasmic APC forms a complex with axin and glycogen synthase kinase-3β (GSK-3β) that promotes β-catenin degradation in the absence of Wnt signaling. Phosphorylation of serine and threonine residues on β-catenin by this complex leads to coupling of β-catenin with ubiquitin, thereby targeting it for destruction. However, activation of Wnt signaling leads to coupling of LDL receptor-related protein with axin and phosphorylation of Dishevelled. Phosphorylated Dishevelled interacts with the APC/axin/GSK-3 complex, thereby inhibiting β-catenin degradation and promoting its accumulation in the cytoplasm. Cytoplasmic β-catenin is then transported to the nucleus where it forms a complex with TCF and lymphoid enhancer factor to promote transcription of several genes involved in cell proliferation and prolonged survival. Loss of APC function prevents destruction of β-catenin, so it accumulates in the cytoplasm and nucleus to drive transcription of genes that represent endpoints of activated Wnt signaling. |
The antitumorigenic effects of APC are not limited to degradation of β-catenin. It also suppresses Wnt signaling by acting on promoters of Wnt responsive genes and shuttling β-catenin out of the nucleus for destruction.14 APC exerts effects through cytoskeletal interactions and chromosomal dynamics. It negatively regulates cell cycling by blocking entry from G0/G1 into the S phase and/or progression through S phase and participates in directed cell migration via binding to microtubules and F-actin.15,16 Finally, APC regulates mitosis by interfering with microtubule dynamics.17 Cells with mutated APC are predisposed to mitotic errors due to defects in mitotic spindles and, thus, have an increased likelihood of aneuploidy.18
Other Molecules Involved in Chromosomal Instability Pathway of Colorectal Carcinoma
Inactivation of APC promotes additional major genetic and chromosomal alterations involving KRAS2 (12p12), genes in chromosome 18q, and TP53 (17q13). Activating KRAS mutations are important to cancer development. This gene encodes a GTP-binding protein that loses GTPase activity when mutated, thereby impairing hydrolysis of active GTP into inactive GDP and resulting in constitutive signaling through the downstream pathway. The original “Vogelgram” depicted KRAS mutations occurring in an intermediate adenoma after APC inactivation (Figure 11.1). Indeed, KRAS mutations can be detected in up to 40% of advanced adenomas (i.e., those with high-grade dysplasia, villous architecture, or size >1 cm), comparable to their frequency in carcinomas. Allelic loss at 18q is found in up to 60% of colorectal carcinomas, affecting the region that contains SMAD2, SMAD4, and DCC.19 Both SMAD2 and SMAD4 are involved in the TGF-β signaling pathway, which is important for growth regulation and programmed cell death. DCC encodes a transmembrane receptor that promotes apoptosis and has been implicated as a key driver of carcinogenesis. However, mutations in DCC and SMAD2 are rare in colorectal carcinoma, whereas SMAD4 may be more influential.20 Indeed, germline SMAD4 mutations are found in a substantial number of patients with juvenile polyposis syndrome, as discussed in Chapter 4. Loss of TP53
 Handling of Colorectal Cancer Resection Specimens
Morphologic Classification of Colorectal Epithelial Tumors
Pathologic Staging Issues: Implementation of the TNM Staging System
Emerging Endoscopic Techniques in the Management of IBD-related Neoplasia
Targeted Therapies for the Treatment of Advanced Colorectal Carcinoma
Surgical Management of Stage IV Colorectal Cancer
Handling of Colorectal Cancer Resection Specimens
Morphologic Classification of Colorectal Epithelial Tumors
Pathologic Staging Issues: Implementation of the TNM Staging System
Emerging Endoscopic Techniques in the Management of IBD-related Neoplasia
Targeted Therapies for the Treatment of Advanced Colorectal Carcinoma
Surgical Management of Stage IV Colorectal Cancer




Related posts:
Handling of Colorectal Cancer Resection Specimens
Morphologic Classification of Colorectal Epithelial Tumors
Pathologic Staging Issues: Implementation of the TNM Staging System
Emerging Endoscopic Techniques in the Management of IBD-related Neoplasia
Targeted Therapies for the Treatment of Advanced Colorectal Carcinoma
Surgical Management of Stage IV Colorectal Cancer
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
