3 Molecular Markers and Pathways in Brain Tumorigenesis
Neoplastic transformation is a multistep process precipitated by the loss of cellular mechanisms controlling cell proliferation and cell–cell interaction. This tumorigenic process involves the interplay between at least two gene classes: oncogenes and tumor suppressor genes (TSGs). Activated oncogenes result in exaggerated cellular signaling that promotes expansion of the aberrant cell population. The TSGs are normal genes that act to inhibit cell proliferation and growth, and inactivation of these genes results in tumor formation or progression. The most common scenario for TSG inactivation results when there is mutation of one allelic copy, followed by loss of all or part of the chromosome bearing the second allele. Consequently, the identification of regions of recurrent chromosomal loss in specific tumor types suggests that a TSG resides in that chromosomal region. These basic themes of oncogene activation and TSG inactivation, coupled with chromosomal loss of heterozygosity, underlie the current molecular understanding of human tumor formation. This chapter reviews the emerging knowledge on the molecular basis of brain tumorigenesis, and discusses primary tumors of the brain as well as other common primary intracranial neoplasms.
 Signaling Pathways Regulating Gliomagenesis
Signaling Pathways Regulating Gliomagenesis
Gliomas are the most common primary neoplasm of the brain, accounting for 40% of all central nervous system (CNS) tumors. The two main types of tumors in the World Health Organization (WHO) classification, astrocytomas and oligodendrogliomas, are named according to their presumed cell of origin and graded according to their histopathological appearance. The system is based primarily on subjective observations of morphological characteristics such as nuclear atypia, mitotic figures, microvascular proliferation, and focal pseudopalisading necrosis. Although tumor grade is currently the most accurate prognostic indicator for clinical outcome, it has been less helpful in determining optimal therapies.
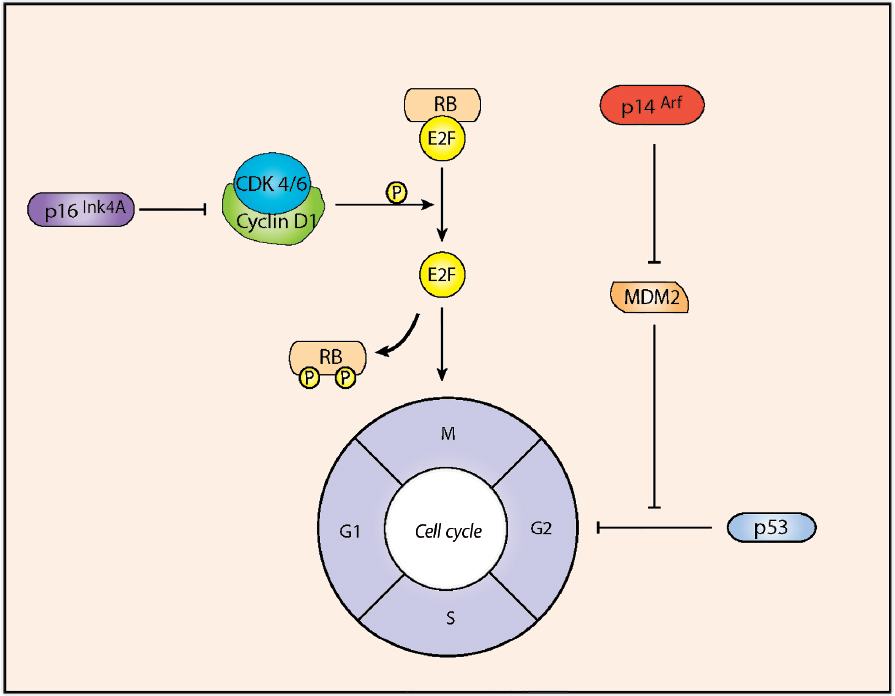
Molecular genetic data gathered since the early 1990s suggest that histologically defined subtypes are even more diverse at a biological level. For instance, the majority of glioblastoma multiforme (GBM) tumors (WHO grade IV) manifest after a short clinical history, thus appearing to arise de novo without clinical or histological evidence of a less malignant precursor lesion. These primary GBMs share mutations affecting two common mechanisms of gliomagenesis: overactive growth factor (mitogen) signaling and disruption of cell cycle control (reviewed below). In contrast, secondary GBMs develop slowly by malignant progression from lower grade gliomas, and the accumulation of genetic alterations over time is a hallmark of these tumors. An unambiguous histopathological distinction of these subtypes has remained elusive, but they clearly evolve through different genetic pathways1,2 (Fig. 3.1).
Special Consideration
• It remains to be shown whether primary and secondary GBMs differ significantly with respect to prognosis, but it appears likely that they will respond differently to specific novel therapies. Ongoing clinical trials and future classification schemes will require incorporation of molecular subtyping.
Growth Factor-Regulated Pathways
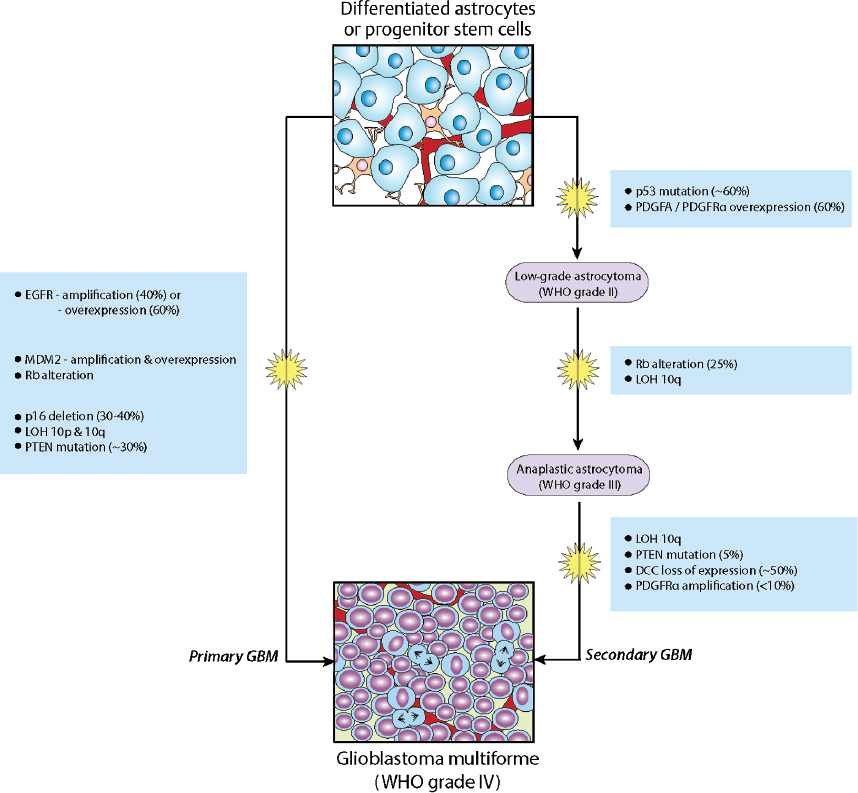
Overactive signaling from several mitogens and their cognate membrane receptors have been implicated in gliomagenesis. Epidermal growth factor (EGF) and its receptor (EGFR), platelet-derived growth factor (PDGF)-A and –B and their respective receptors (PDGF-α, -β), transforming growth factor-α (TGF-α) acting through EGFR, and insulin-like growth factor-I (IGF-I) and its receptor (IGFR) are often involved in stimulating tumor cell proliferation.3,4 Frequently, components of these mitogenic pathways are overexpressed in gliomas, or their upstream receptors harbor constitutively active mutations.5
Epidermal growth factor receptor amplifications and mutations have long been recognized as common (40%) gain of function alterations in GBMs.3,6 Polymorphism in the 5′-untranslated region of EGF has also been implicated in gliomagenesis.7 Patients with the –GA/–GG genotypes had higher levels of EGF within tumor tissue, irrespective of EGFR status, and had significantly shorter overall progression-free survival, compared to the common –AA genotype.
Three key signaling pathways downstream of aberrant growth factor/receptors involve Ras-Raf-MAPK (mitogen activated protein kinase), PI3K/Akt-PKB (phosphoinositide 3’kinase/Akt-protein kinase B), and PLC-γ/PKC (phospholipase C-γ/protein kinase C) (Fig. 3.2). These signaling pathways regulate cellular processes including proliferation, differentiation, and apoptosis, and have traditionally been thought to influence nuclear factors to alter cellular transcription and hence potential transformation.8 However, both the Ras and PI3K pathways primarily alter existing growth-promoting messenger RNA (mRNA) to become associated with polysomes and hence actively transcribed.9 The PI3K/Akt-PKB pathway, in particular, has received much attention given its negative regulation by the tyrosine phosphatase/tensin (PTEN) homolog protein,10 the most common loss of functional genetic alteration in GBMs. Additionally, the PI3K/Akt-PKB pathway modulates, through intermediate signal transducers, EGFR-regulated activation of the transcription nuclear factor (NF)-κB involved in inflammatory and transformation pathways.11,12
Special Consideration
• Study findings justify targeting the EGF/EGFR-regulated pathways and suggest that a single nucleotide polymorphism in EGF may be a useful marker for identifying patients with poor prognosis and who may benefit from anti-EGF/EGFR targeted biological therapies.
The Ras-Raf-MAPK signaling pathway is known to transduce mitogenic signals in a variety of cells, including astrocytomas. Ras is a small (21 kd) intracellular protein that exists bound to a guanine nucleotide (such as guanosine diphosphate, GDP) in its inactive state. Upon delivery of an appropriate stimulus, its intrinsic guanosine triphosphatase (GTPase) activity is upregulated through a series of intermediary adapter molecules. Accumulation of Ras–guanosine triphosphate (GTP) ultimately leads to increased expression or repression of specific target genes.
Ras activation is implicated in other key processes such as cell adhesion through activation of integrin-mediated focal adhesion kinase (FAK) in astrocytomas.13 The GBMs are known to overexpress FAK, which, if exogenously expressed in GBM cells, activates Ras, presumably by interaction of FAK with adapter molecules such as Shc. Mutations of other small-GTPase members of the Ras superfamily, including Rac1, have also been linked to cytoskeletal alterations and invasion of glioma cells.14 Rho, a related GTPase family member, has also been implicated in cell shape and motility, with recent evidence that inhibition of Rho leads to radiosensitization.15 Significantly, expression of p190Rho-GAP, the endogenous inactivator of Rho, inhibits PDGF-mediated oligodendroglioma development.16
Pearl
• Loss of PTEN function results in an overactive, unregulated Akt/PKB oncoprotein that inhibits apoptosis and promotes cell proliferation.
Cell Cycle–Regulated Pathways
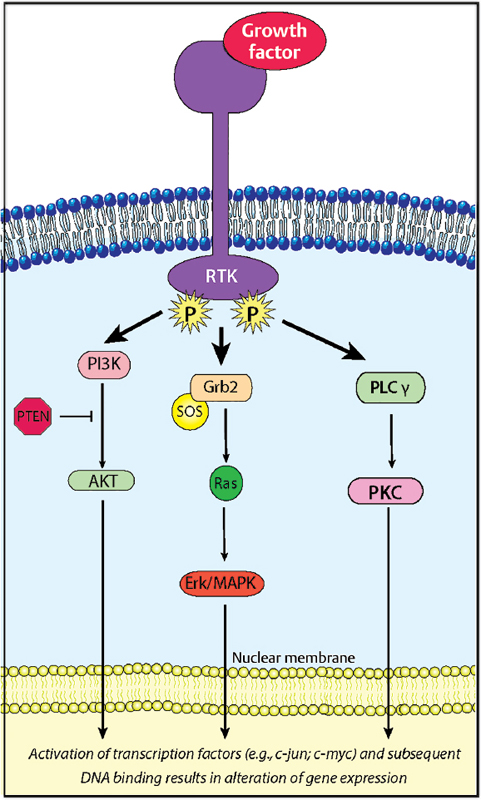
A variety of signaling pathways converge at various checkpoints to tightly regulate progression through the cell cycle. Alterations in the retinoblastoma (Rb)- and p53- dependent pathways are implicated in glial transformation through control of progression at the G1 and G2 checkpoints.17,18 These pathways are regulated by the tumor suppressor proteins p16INK4a and p14ARF, respectively, which are encoded by the INK4a-ARF (inhibitor of cyclin-dependent kinase 4a–alternative reading frame) locus.19
The retinoblastoma (Rb) gene was initially identified as a tumor suppressor and, within the CNS, Rb null mice demonstrate aberrant cell division and apoptosis, revealing a role in cell proliferation and survival.20 Rb mutations have been found in about 20% of WHO grade III gliomas, thus supporting a role in their formation.21,22 Low-level phosphorylation of Rb by cyclin-dependent kinase 4/6 (CDK4/6)-cyclin D1 complexes disrupts interactions between Rb and E2F, which relieves Rb-mediated repression of E2F-target genes required for cell cycle progression.22 Loss of the tumor suppressor p16INK4a results in deregulated Rb phosphorylation and, consequently, in unchecked G1-S progression (Fig. 3.3).
The p53 transcription factor activates target genes that promote cell cycle arrest or apoptosis in response to cellular stresses such as oncogene activation.23 The activity of p53 is regulated by the interplay between MDM2 (murine double minute 2)-mediated degradation and p14ARF-mediated inhibition of MDM224 (Fig. 3.3). Over two thirds of GBMs and anaplastic astrocytomas exhibit deregulated G1-S transition associated with inactivation of the p53 pathway, with either mutation of p53, amplification of MDM2, or homozygous deletion/mutation of p14ARF.25
Pearl
• Sixty percent of GBMs have deletion or inactivation of the INK4a-RAF locus.
 Medulloblastomas
Medulloblastomas
Deregulated Developmental Signaling in Medulloblastomas
Medulloblastoma (MB), the most common malignant brain tumor of childhood, is a primitive neuroectodermal tumor arising from the granule cell progenitors of the cerebellum.26 It accounts for up to 40% of all posterior fossa neoplasms.26 Medulloblastoma is currently diagnosed on histopathological examination of tumor tissue; however, recent integrated genomic approaches have demonstrated that MB is composed of multiple clinically and molecularly distinct subgroups.27,28 Emerging evidence indicates that the different precursor cell populations that form the cerebellum and the cell signaling pathways that regulate its development likely represent distinct compartments from which the subtypes of MB arise.
The main limitation in current therapies for MB is the lack of specificity of drugs used. Advances in understanding the molecular mechanisms have provided clues on the pathogenesis of MBs and could substantially improve the management of these neoplasms by employing targeted treatments. At least four subgroups of MB have now been described—Wnt, Shh, Group 3, and Group 4—and there may be other subtypes within these subgroups.29
Sonic-Hedgehog-Patched Signaling
The Sonic Hedgehog (Shh)-Patched (Ptch) signaling pathway is a major mitogenic regulator of cerebellar granule cell progenitors (CGCPs).30,31 During cerebellar development, the glycoprotein Shh is primarily synthesized and secreted by pyramidal neurons and upon binding to the receptor Ptch, is mainly expressed on granule precursor cells, which activates the pathway by relieving Smoothened (Smoh)-mediated inhibition (Fig. 3.4). Disruption of Ptch activity causes aberrant Smoh activation of downstream target genes, such as the Gli family of transcription factors, to induce oncogenesis. Overexpression of Shh or Gli leads to oncogenic transformation.32,33
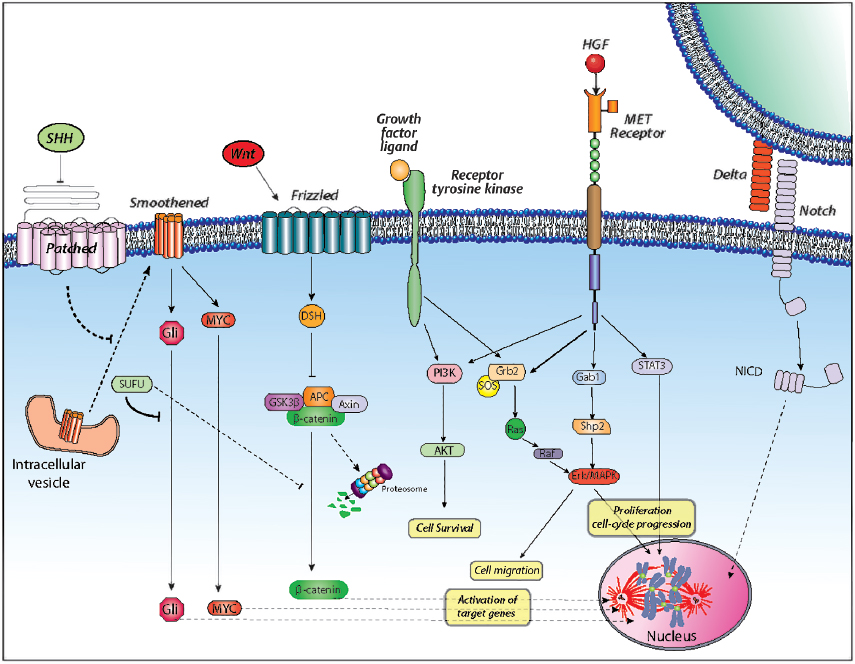
The crucial role of this pathway in progenitor proliferation has been linked to cell-cycle control at the G1 checkpoint, wherein Shh induces Cyclin D1 and Cyclin D2 expression, Rb hyperphosphorylation, and E2F activation.34
A subset of MB patients was shown to express mutated SUFU (suppressor of fused),35 a protein originally isolated as a downstream repressor of Shh signaling that functions by exporting Gli proteins from the nucleus for degradation. SUFU mutants that fail to export Gli proteins from the nucleus result in aberrant Shh signaling. Therefore, MB can arise from deregulation of Shh-Ptch signaling in granule cell precursors via mutations of various pathway components. Mutations affecting PTCH1 and other components of the Shh signaling complex, namely SUFU, PTCH2, and SMO, have been identified in up to 25% of sporadic cases of MB.35,36
Pearl
• The role of PTCH1 mutations in the genesis of MB is supported by murine models, where approximately 14% of mice with a heterozygous deletion of Ptc (Ptc+/2) develop MB by 10 months of age, with peak tumor incidence occurring between 16 and 24 weeks of age.
• Many Gorlin’s syndrome (or nevoid basal cell carcinoma syndrome) patients bear germline mutations in PTCH and have increased risk of developing MB. The PTCH gene is located on chromosome 9q, and mutations in that region are also found in 10 to 20% of spontaneous MBs. SUFU is localized to chromosome 10q, and about 40% of MBs carry abnormalities in this region. Overexpression of the polycomb transcription regulator BMI-1 has been reported in a subset of human primary MBs and linked with Shh pathway activation. This gene regulates CGCP proliferation during cerebellar development, and its correlation with Shh pathway activity suggests an alternative mechanism for Shh signaling in the development of MB.
Wnt Signaling
The Wnt signaling pathway has been well studied in multiple developmental systems and has been implicated in tumorigenesis of many tissues.37
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


