Key points
- 1.
Malignant cells develop from molecular abnormalities in growth control.
- 2.
The molecular abnormalities are in one or more of 10 key pathways.
- 3.
These altered pathways provide important targets for new therapeutic agents.
- 4.
Angiogenesis, PARP, immunologic cascades and cellular signaling pathways are examples of important targets.
- 5.
Antibody drug conjugates are new therapeutic agents which rely of cell surface targets.
- 6.
Many targeted agents have unique toxicity profiles.
- 7.
DNA methylation is a new and novel target.
Biology of gynecologic cancers
Gynecologic cancer remains a major health problem for women ( Tables 16.1 and 16.2 ). There is great heterogeneity that exists within gynecologic malignancies. Across the different tumor sites, there is a wide variety of clinical sequelae due to considerable differences on a genomic, molecular, and pathologic level. For example, endometrial cancer (EC) has classically divided tumors into Type I and type II cancers. Type I tumors are estrogen-driven and commonly demonstrate endometrioid histology. Type II ECs are estrogen-independent and are represented by non-endometrioid histologies, such as serous, clear cell, and carcinosarcoma. The two distinct categories of EC exhibit different malignant behaviors and have different clinical presentations, prognosis, and responses to treatment. Similar stratification based on a histology is seen in epithelial ovarian cancer (EOC). Within EOC there are five distinct histologic subtypes, including high-grade and low-grade serous ovarian cancer, endometrioid, mucinous, and clear cell. The five tumor subtypes possess diverse morphologic features, and successful histopathological diagnosis is key to determining treatment. In gynecologic cancers, there has been a shift from relying on histologic features for diagnosis to characterizing these tumors on a molecular level, providing a more exact classification system based on genomic analysis. These findings have been extracted from the Tumor Cancer Genome Atlas (TCGA). Further, studies focused on these molecular changes have led to development of numerous targeted therapies for gynecologic cancers.
| 1980 | 1990 | 2000 | 2010 | 2017 | |
|---|---|---|---|---|---|
| Cervical | 12.8 | 10.7 | 7.7 | 6.8 | 6.3 |
| Endometrial | 27.3 | 25.0 | 24.8 | 27.5 | 28.6 |
| Ovarian | 15.5 | 15.4 | 14.4 | 12.9 | 10.2 |
| 1980 | 1990 | 2000 | 2010 | 2017 | |
|---|---|---|---|---|---|
| Cervical | 4.5 a | 3.7 | 2.8 | 2.3 | 2.2 |
| Endometrial | 5.1 | 4.3 | 4.1 | 4.5 | 5.0 |
| Ovarian | 9.3 | 9.3 | 8.9 | 7.8 | 6.6 |
a Rates presented as per 100,000 women (Data from Surveillance, Epidemiology, and End Results [SEER] 18 registries, National Cancer Institute, 2020).
Molecular changes in gynecologic malignancies
There exists a highly regulated and complex interaction between intracellular functions and environmental factors that is essential for normal cellular growth and death. There are a multitude of mechanisms designed to maintain homeostasis within these processes. However, an accumulation of molecular alterations allows for cell growth to escape these protective mechanisms, leading to malignant transformation. On a genetic level, overexpression of oncogenes or deletion of tumor suppressor genes are common aberrations that occur. Beyond this, there are several complex processes that can be altered which also lead to the development of cancer. Hanahan and Weinberg described these capabilities that are acquired by normal cells as the “hallmarks of cancer” ( Fig. 16.1 ). These processes include promotion of self-sustained growth via over-production of cellular growth signals and angiogenesis. Furthermore, cancerous growth is marked by evasion of antigrowth signals, such as apoptosis, allowing cells to grow even in the presence of cellular damage. The ability for tumor cells to metastasize and invade new tissue is key to continued growth once resources and space become restricted at a previously invaded site.
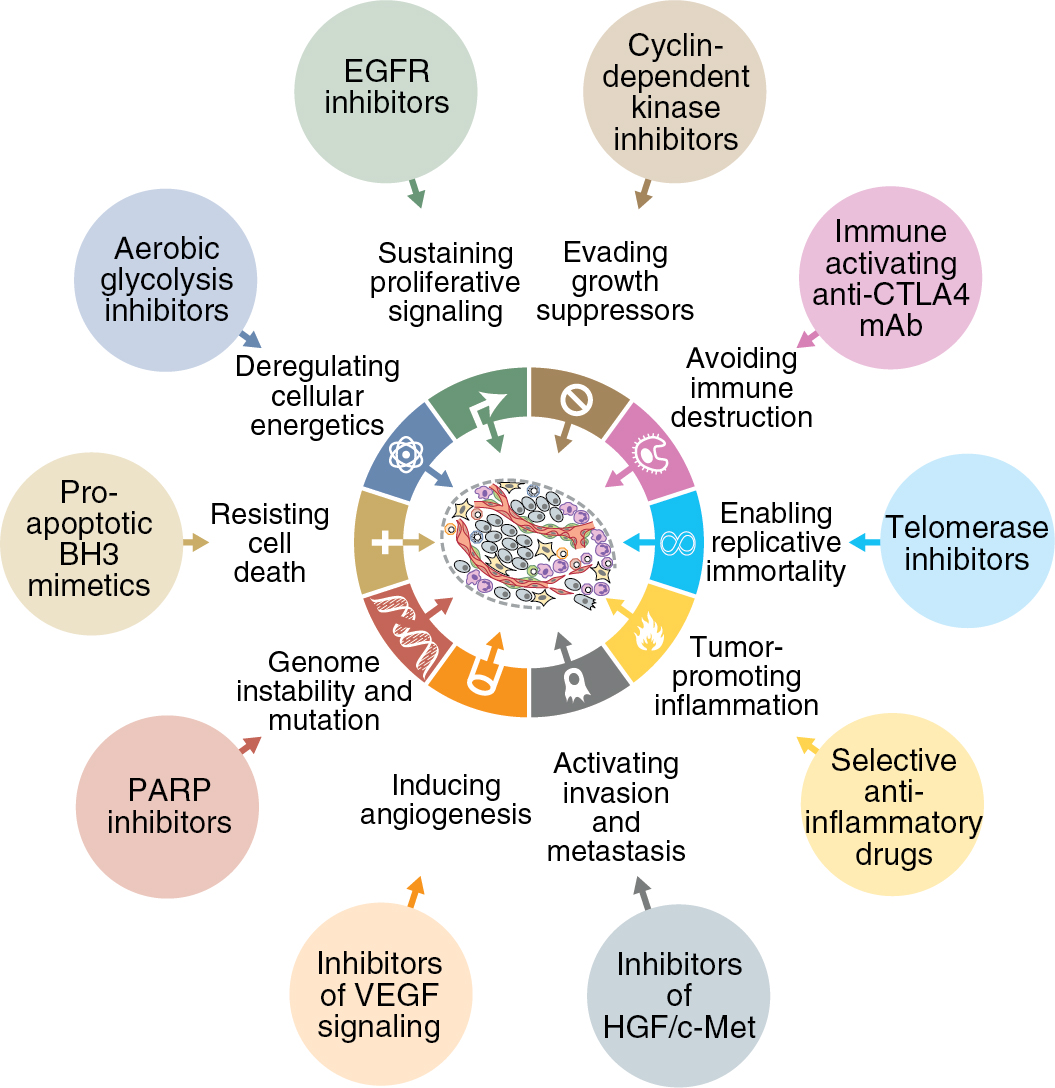
The acquisition of these survival capabilities is due to dysfunction of normal pathways of cellular growth, and it is changes in these pathways that lead to cancer progression. Identifying these changes has become the focus for development of new and novel cancer therapeutics. For decades, treatment for gynecologic cancers has been relegated to either cytotoxic chemotherapy or radiation therapy. While these treatments are effective at achieving tumor reduction, they do not discriminate well between normal cells and tumor cells. This leads to side effects secondary to the non-specific nature of these therapies. In particular, rapidly dividing cells, such as cancer cells, are most sensitive to these treatment modalities, and a majority of side effects are seen in normal host cells that also have high turnover rates, such as host bone marrow, the gastrointestinal tract, and the integumentary system. Targeted therapies are meant to provide an alternative approach that inhibits cancer growth and reduces tumor burden via a different mechanism than cytotoxic or radiotherapy, while avoiding damage to host cells by providing a more specific mechanism. The development of targeted therapy focuses on pathways that are altered during tumorigenesis. Targeted therapeutics take advantage of these sentinel changes in malignant transformation in order to provide treatment specific to the cancer cell, while sparing normal cells. In order to identify effective targeted therapies, it is important to have a thorough understanding of the molecular changes that produce abnormal cellular growth in cancer cells. There are a wide variety of changes that occur on a molecular level within all cancers; however, there are some key pathways that are commonly altered, specifically within gynecologic malignancies, that are essential to the biology of these cancers and how to treat them.
Cell cycle
The cell cycle is a highly regulated and complex process that governs cellular division. In normal cells, there are multiple checkpoints that ensure proper progression through this process and allow for the correction of DNA mutation prior to advancing to the next step or lead to cell death to prevent error-mediated growth. The progression through the cell cycle is regulated by proteins called cyclin-dependent kinases (CDK). These proteins are a family of serine and threonine kinases that are activated at specific points throughout the cell cycle via signaling from a variety of regulatory proteins ( Fig. 16.2 ). Alterations in CDK activity can lead to unregulated growth, genomic instability, and ultimately, malignant transformation. Opposing the CDK activity are a group of proteins called CDK inhibitors, and alterations of their activity can also lead to malignant transformation. The origin of gynecologic malignancies can be linked to specific cell cycle alterations. The activity of tumor suppressor or oncogenes can be altered and lead to dysfunction in the cell cycle. In human papilloma virus (HPV)-associated malignancies, such as cervical, vulvar, or vaginal cancers, these are secondary to effects from viral oncogenes E6 and E7 on tumor suppressor proteins p53 and pRb, respectively. Additionally, the occurrence of a somatic mutation in TP53 is a sentinel event in the establishment of high-grade Serous Ovarian Cancer (HGSOC), as well as serous uterine cancer. Therapeutics aimed at blocking CDK activity in gynecologic malignancies are actively being studied as monotherapy and in combination with other treatment modalities commonly used.
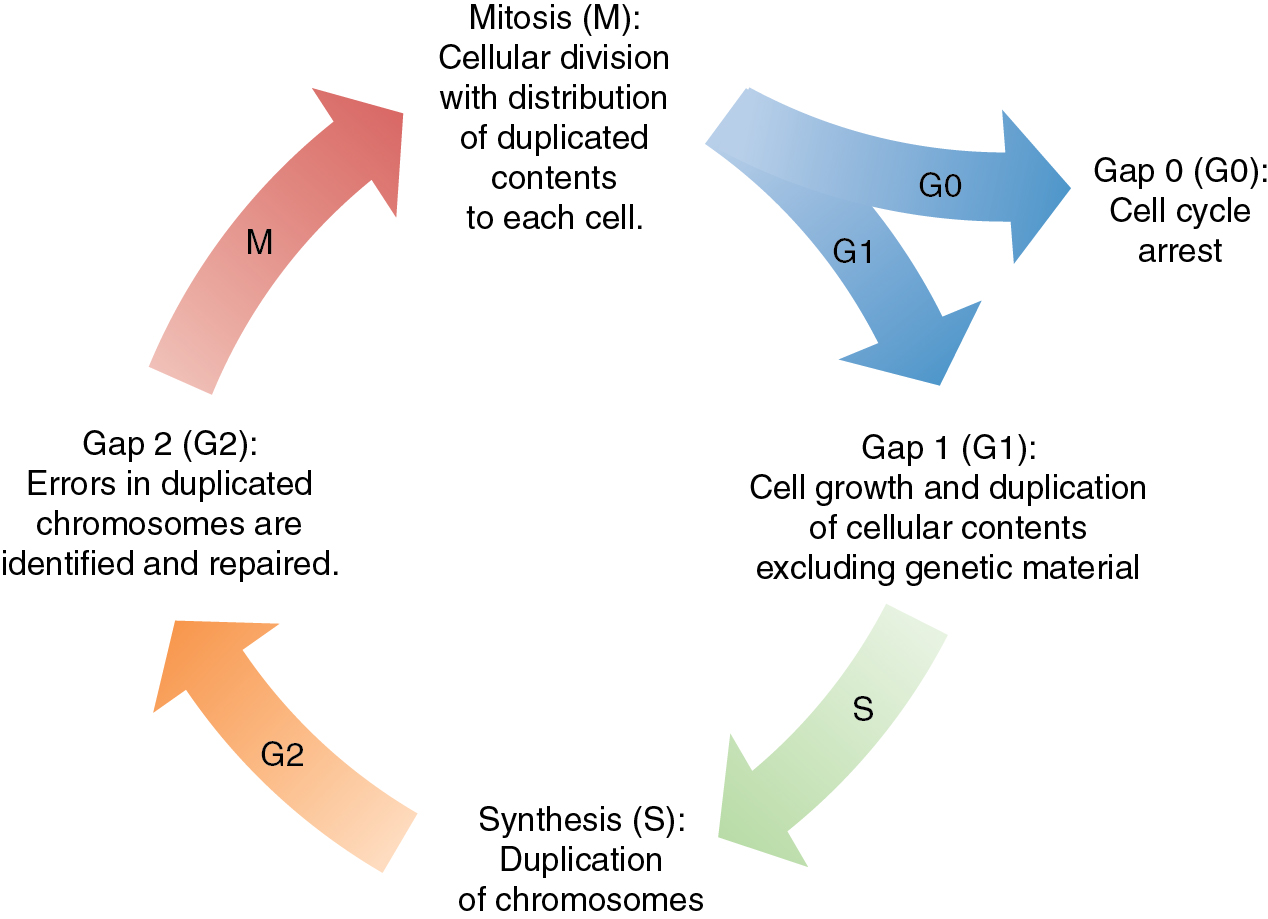
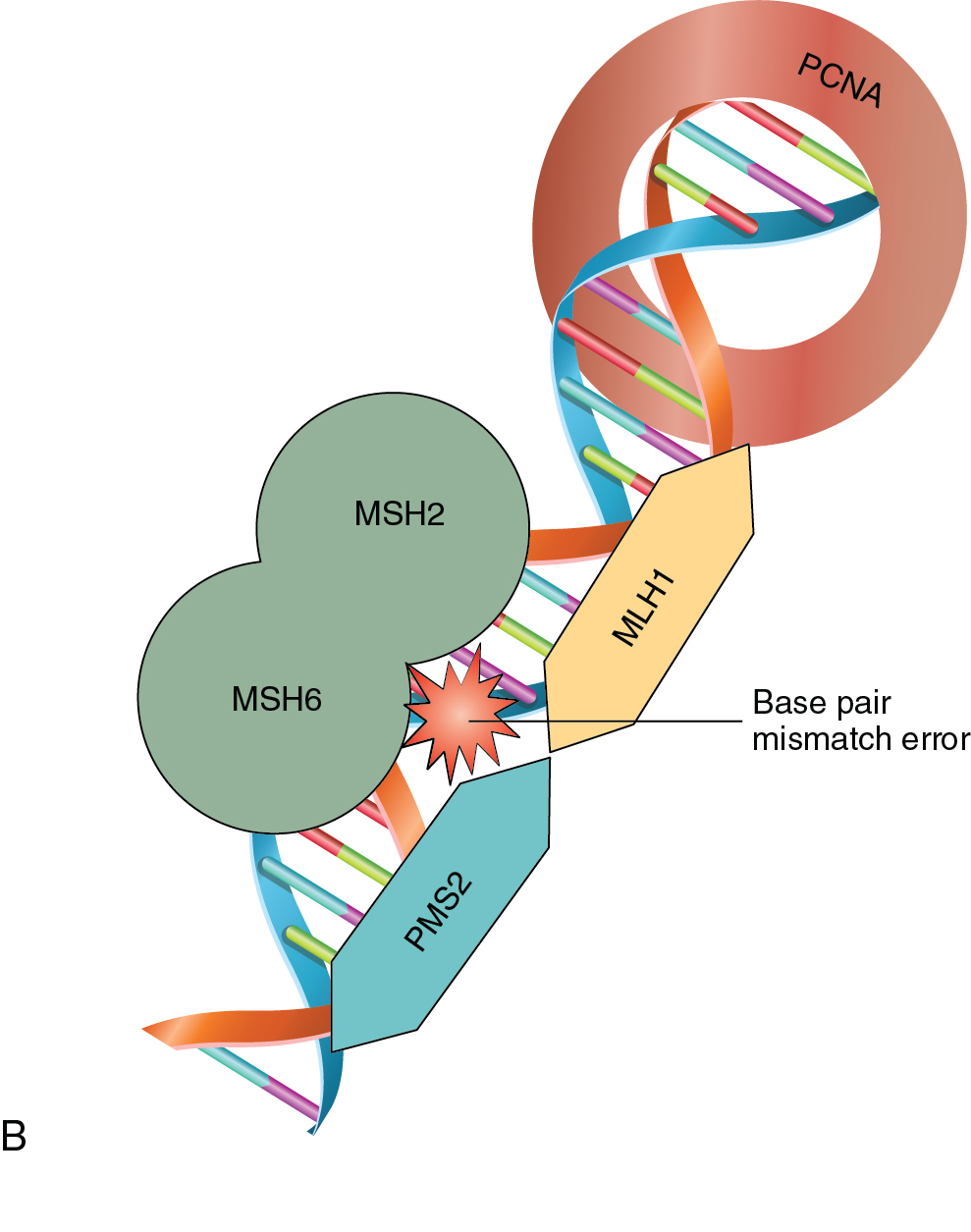
As a normal cell progresses through the cell cycle, a key process to successful cellular division is the replication of genetic material. DNA replication occurs in the S-phase of the cell cycle where genetic material is doubled prior to the cell dividing with equal distribution of DNA into each cell. DNA replication is a complex process that is prone to error. There are inherent mechanisms in place to identify and correct any errors in this process. There are also built-in systems that mark a cell for destruction in the event that errors are beyond correcting. Unchecked mistakes in DNA replication are key drivers in cancer development. Defects in genetic stability leading to replicative mutations and subsequent clonal expansion lead to tumorigenesis and cancer progression. Mutations in genes integral to maintaining the fidelity of DNA replication are associated with human cancer syndromes. Two commonly encountered gynecologic cancer syndromes are associated with mutations in breast cancer gene 1 and 2 (BRCA1 and BRCA2) and other genes of the homologous recombination (HR) repair pathway, and errors in DNA mismatch repair (MMR) ( Fig. 16.3 A and B).
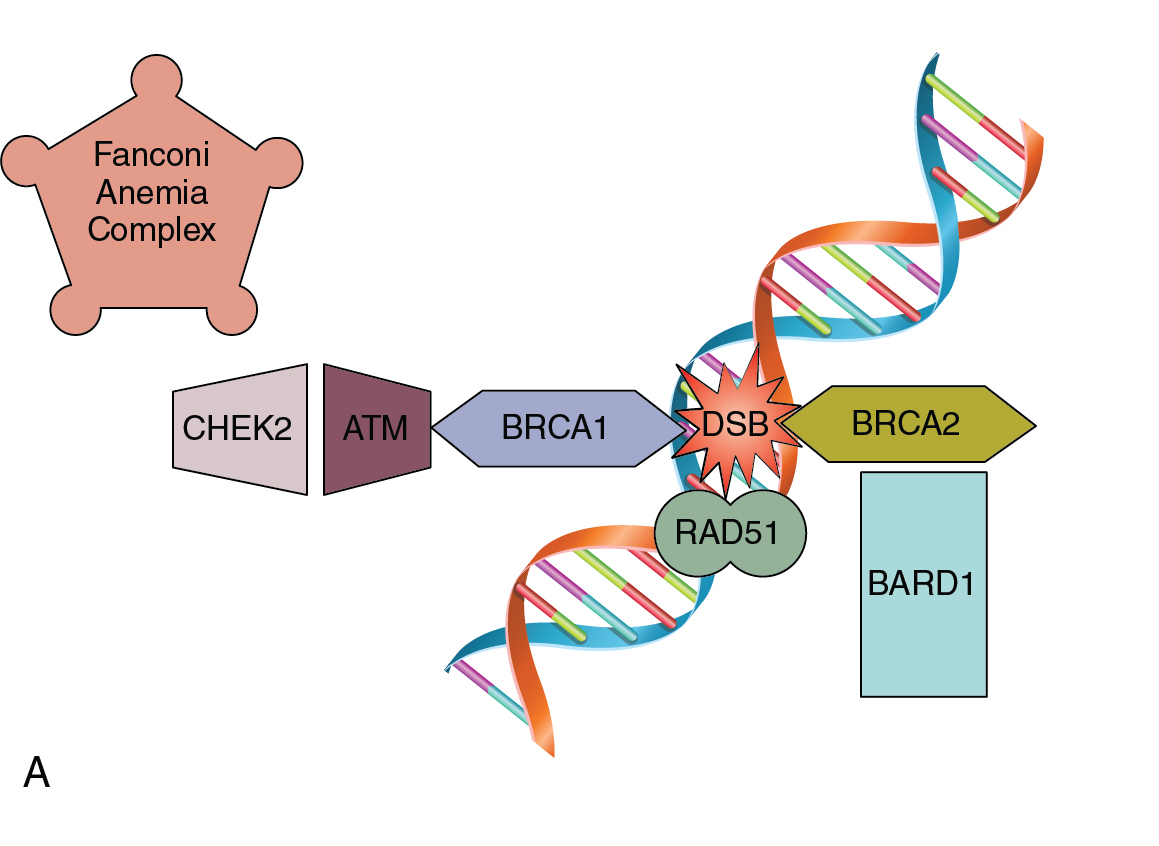
Pathogenic mutations in BRCA 1 and 2 are associated with a constellation of various malignancies. These genes play an integral role in the HR repair pathway and function to correct double-stranded DNA breaks. Germline mutations in HR genes such as BRCA 1/2 increase the lifetime risk of certain malignancies, such as ovarian cancer. Approximately 16% of ovarian cancers are attributable to inheritable mutations in genes such as BRCA 1/2. Women with mutations in BRCA 1 and 2 genes, respectively, have a 15% and 40% lifetime risk of developing ovarian cancer. The BRCA status of a patient diagnosed with ovarian cancer has implications for their family members, as well as prognosis and treatment options for individual patients. Currently, the recommendation is to perform genetic testing on all patients with non-mucinous epithelial ovarian malignancies. Mutations in other genes within the Fanconi Anemia pathway, including partner and localizer of BRCA2 (PALB2), BRCA1 interacting protein 1 (BRIP1), and RAD51 paralog C (RAD51C) v, are inheritable mutations that also increase a female’s risk of developing ovarian cancer and have similar tumor behavior and response to therapy. Additionally, HR deficiency can occur due to somatic mutations within the tumor, which are not inheritable changes, but are identical to ovarian cancers in patients with germline BRCA mutations. Genes related to HR, such as RAD51, checkpoint kinase 2 (CHEK2), ataxia telangiectasia mutated (ATM), and BRCA1 associated ring domain 1 (BARD1), when altered, cause defects in DNA repair leading to genomic instability that can be measured as loss of heterozygosity (LOH), which can subsequently be used as a marker for HR deficiency. These genetic mutations create changes within the tumor that present potential opportunities for additional therapy via poly (ADP-ribose) polymerase (PARP) inhibitors via a mechanism termed “synthetic lethality” by blocking both single-stranded and double-stranded DNA repair mechanisms within tumor cells that are HR deficient.
In the 1960s, Dr. Henry Lynch and colleagues described an autosomal-dominant tumor syndrome that predisposes individuals to an increased risk of developing a variety of tumors. This tumor syndrome was named Lynch syndrome, after the researcher, and is also known as hereditary non-polyposis colon cancer (HNPCC). In addition to a significantly increased risk of acquiring colon cancer at a young age, women with Lynch syndrome have a 40% to 60% risk of endometrial cancer, as well as a 12% risk of ovarian cancer. Lynch syndrome is caused by mutations in genes associated with DNA MMR – MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), MutS homolog 6 (MSH6), and PMS1 homolog 2 (PMS2). In patients with gynecologic malignancies where there is concern for Lynch syndrome, evaluation by immunohistochemistry (IHC) to detect loss of MMR proteins should be performed. Absent staining for these proteins would then lead to germline testing for potential inheritable mutations. Furthermore, absence or loss of function of these proteins has therapeutic implications for endometrial cancer patients. Errors in MMR can lead to a phenomenon known as microsatellite instability (MSI) which, if found to be present in gynecologic cancers, serves as a marker predictive of response to certain therapies such as immunotherapy.
Agents
Mechanism of targeted agents
There are a variety of mechanisms to deliver targeted therapy in an effort to inhibit tumor progression. Monoclonal antibodies (mAb) are biological molecules that bind cancer-associated molecules. This includes cell surface molecules as well as ligands. Cancer cells, along with cells and molecules within the tumor microevironment (TME) are potential targets of mAb. Monoclonal antibodies are classified based on the origin of the antibody. Common sources for mAb include human and murine sources, as well as their combination in the form of chimeric monoclonal antibodies. The source of the antibody is relevant to the possible immune response to the therapy in humans. Intuitively, mAb with a larger portion derived from a human has a lower potential for response than those that are more murine. An important property of mAbs is that they are highly specific for a single target. The antibody structure allows for high variability, allowing for design of an mAb that is unique to single antigen target. This helps minimize unintended off-target effects from mAb therapy.
Another mechanism of targeted agents are small molecule inhibitors. One common target of small molecule inhibitors is blockage of tyrosine kinase activity. Conventional tyrosine kinase molecules are transmembrane proteins, called receptor tyrosine kinase, that are the catalyst of a wide array of cellular functions. The fundamental activity of tyrosine kinase enzymes includes a signaling via binding to the extracellular domain and activation of kinase activities which utilizes adenosine triphosphate (ATP) to transfer a phosphate group from the cytoplasmic component to a downstream target. The transfer of the phosphate group results in changes to the receptor protein which leads to a distinct biologic function within the pathway. In non-receptor tyrosine kinases, similar activation of kinase activity followed by phosphate transfer takes place intracellularly within the cytoplasm or the nucleus. These are typically activated through interaction with a thorough extracellular process. Small molecule inhibitors are designed to block tyrosine kinase activities that are associated with processes promoting malignant transformation and growth. As opposed to the sustained half-life of mAbs, small molecule inhibitors have short half-lives and are administered as frequent oral doses. The typical mechanism of the small molecule inhibitor takes advantage of the high level of fidelity found at the ATP-binding domain in tyrosine kinase enzymes. Blockage of this specific site within a single enzyme within the molecular pathway provides anti-tumor activity and allows for flexibility in designing therapies to target a variety of pathways by inhibiting tyrosine kinase molecules that are the crux of cancer promoting pathways.
Antibody drug conjugates (ADCs) are a rapidly expanding classification of cancer therapy that utilize immunoglobulins to deliver cytotoxic therapy to tumor cells. The antibody component binds to tumor-specific antigens in an effort to limit host toxicity. ADCs carry potent small-molecule drugs to target key molecular pathways within tumor cells and the tumor microenvironment. The linker between the tumor-specific antibody and the toxic payload is an important component of ADC functionality. Linkers modulate the pharmacokinetics of ADCs, which determines the efficacy and potential toxicity of the treatment. The linker is meant to be stable enough to prevent premature drug delivery as well as maintain the association between the antibody and the cytotoxic therapy while in solution. Additionally, the linker within ADCs can adjust the molecular properties of the drug facilitating intracellular delivery.
There are a wide variety of other agents that are currently being investigated as targeted therapy for gynecologic malignancies. As our understanding of malignant molecular processes continues to grow, the advancement of targeted agents will progress, improving our ability to deliver cancer treatment while minimizing side effects.
Targets
Angiogenesis
The generation of new blood vessels, angiogenesis, facilitates the delivery of nutrients, oxygen, and other growth factors to tissues. This is a normal process that contributes to wound healing and tissue growth. However, malignant tumors possess the ability to induce angiogenesis and increase blood supply to the tumor itself. This provides the necessary resources to allow the tumor to rapidly grow. The tumor microenvironment demonstrates the overexpression of pro-angiogenic growth factors which leads to sustained vascular development. Angiogenesis is a process that is balanced by both pro- and anti-angiogenic factors, such as vascular endothelial growth factors (VEGF), platelet derived endothelial growth factor, interleukin-8, and other proteins. Tumor cells express growth factors that promote the balance towards pro-angiogenesis and neovascularization. Angiogenesis within the tumor microenvironment is a rushed and imperfect process in order to keep up with the demands of the growing tumor. These vessels are more irregular in both morphology and integrity. The growth factors that regulate vascular growth work via interaction with endothelial cells, which line the blood vessels. In malignant growth, these endothelial cells are triggered to rapidly divide as new vessels are being generated.
Targeting angiogenesis is an effective strategy in cancer treatment. In gynecologic oncology, anti-angiogenics represent the first targeted therapy to be utilized in the treatment of these cancers. Strategies to inhibit angiogenesis include targeting both VEGF and VEGF receptor activity as well as inhibition of downstream signaling in angiogenesis processes ( Table 16.3 ).
| GOG 213 | GOG 218 | ICON 7 | OCEANS | AURELIA | PAOLA | |
|---|---|---|---|---|---|---|
| Total Patients | 674 | 1873 | 1528 | 484 | 361 | 806 |
| Study Arms |
|
|
|
|
| |
| Primary Endpoint | OS | OS | PFS | PFS | PFS | PFS |
| Secondary Endpoints | PFS, QoL, Hypersensitivity | Toxicity, QoL, Translational data | OS, RR, QoL | ORR, OS, DoR | ORR, OS, QoL | PFS2, OS, TSST, QoL |
| Primary or Recurrent Disease | PS Recurrent | Primary | Primary | PS Recurrent | PR Recurrent | Maintenance |
a Investigator’s choice chemo options: Paclitaxel 80 mg/m 2 day 1, 8, 15, 22 q4weeks; Topotecan 4 mg/m 2 day 1, 8, 15 q4weeks; Pegylated Doxorubicin 40 mg/m 2 q4weeks.
Vascular endothelial growth factors and receptors
The VEGF pathway is the key regulatory process in angiogenesis. The VEGF pathway includes seven different ligands which can bind to three different VEGF receptors ( Fig.16.4 ). These receptors are tyrosine kinase receptors typically found on the membranes of endothelial cells lining vessels. However, tumor cells are able to express VEGF receptors on their cell surface as well. Activation of the VEGF pathway can lead to signaling of other important oncologic molecular pathways, including phosphoinositide-3-kinase/protein kinase B pro (PI3K/AKT), mitogen-activated protein kinase (MAPK) pathway, focal adhesion kinase (FAK), and v-src sarcoma viral oncogene. The overexpression of VEGF has been shown in a wide variety of tumors including gynecologic cancers. There is a direct correlation between the overexpression of VEGF within the tumor microenvironment and tumor progression which portends a worse prognosis. VEGF activity is an important contributor to sustained tumor growth, and targeted anti-angiogenesis therapies have demonstrated success as treatment for gynecologic cancers.
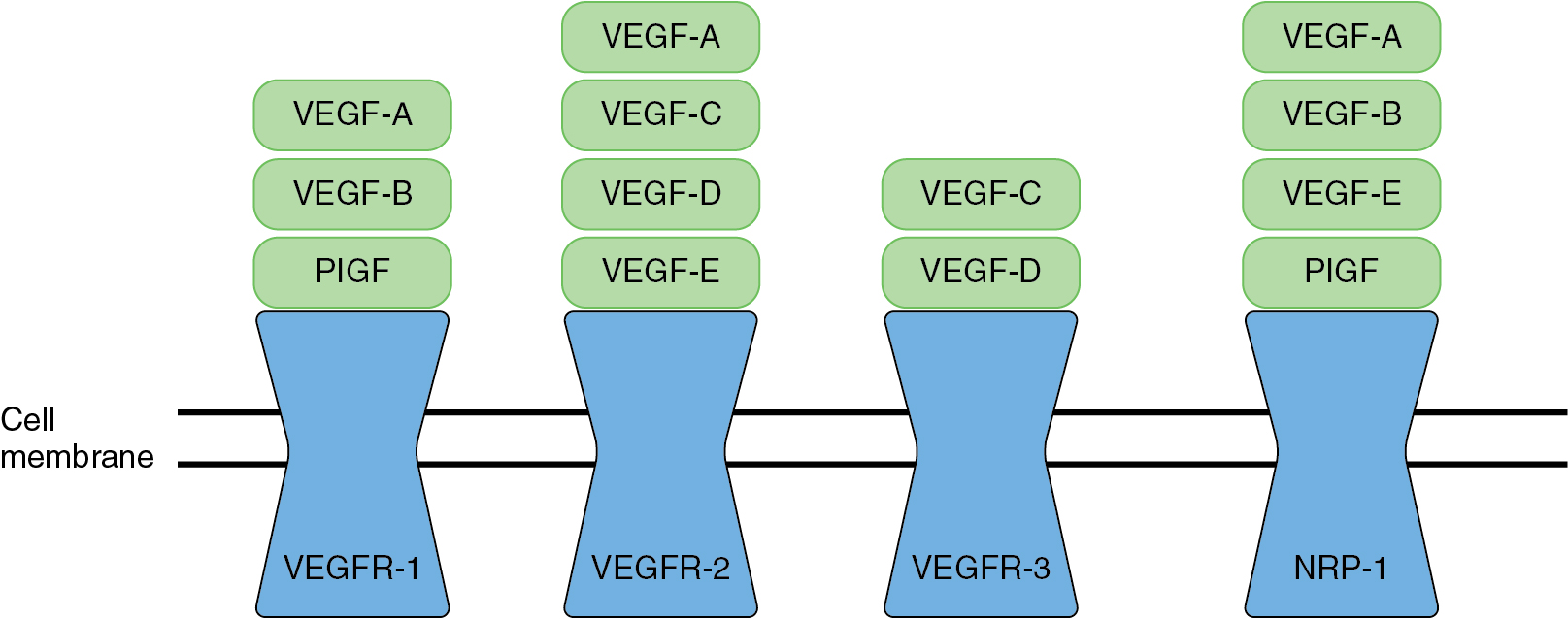
Agents that target VEGF-dependent angiogenic pathways are described in detail in the chapter 15 (Targeted Therapy).
The PI3K/AKT pathway ( Fig. 16.5 ) is integral to cellular growth, homeostasis, and genomic stability. Aberrant signaling in this pathway is a common contributor to cancer development and progression as signaling via this pathway contributes to chemoresistance, angiogenesis, and cell survival. Tyrosine kinase receptor and G-protein coupled receptors provide the primary signaling transduction for this pathway through interactions with various cytokines and growth factors. The pathway begins with activation of PI3K proteins which in turn leads to phosphorylation of phosphatidylionositol-4,5-bisphosphate (PIP2), forming phosphatidylionositol-3,4,5-triphosphate (PIP3). PIP3 activates various intracellular targets including AKT, which is an important mediator of a variety of cellular processes. Mammalian target of rapamycin (mTOR) is a serine/threonine kinase which is activated by AKT signaling. Upregulation of mTOR1 leads to activation of protein S6 kinase which promotes progression of the cell cycle from G1 to S-phase and cellular growth.
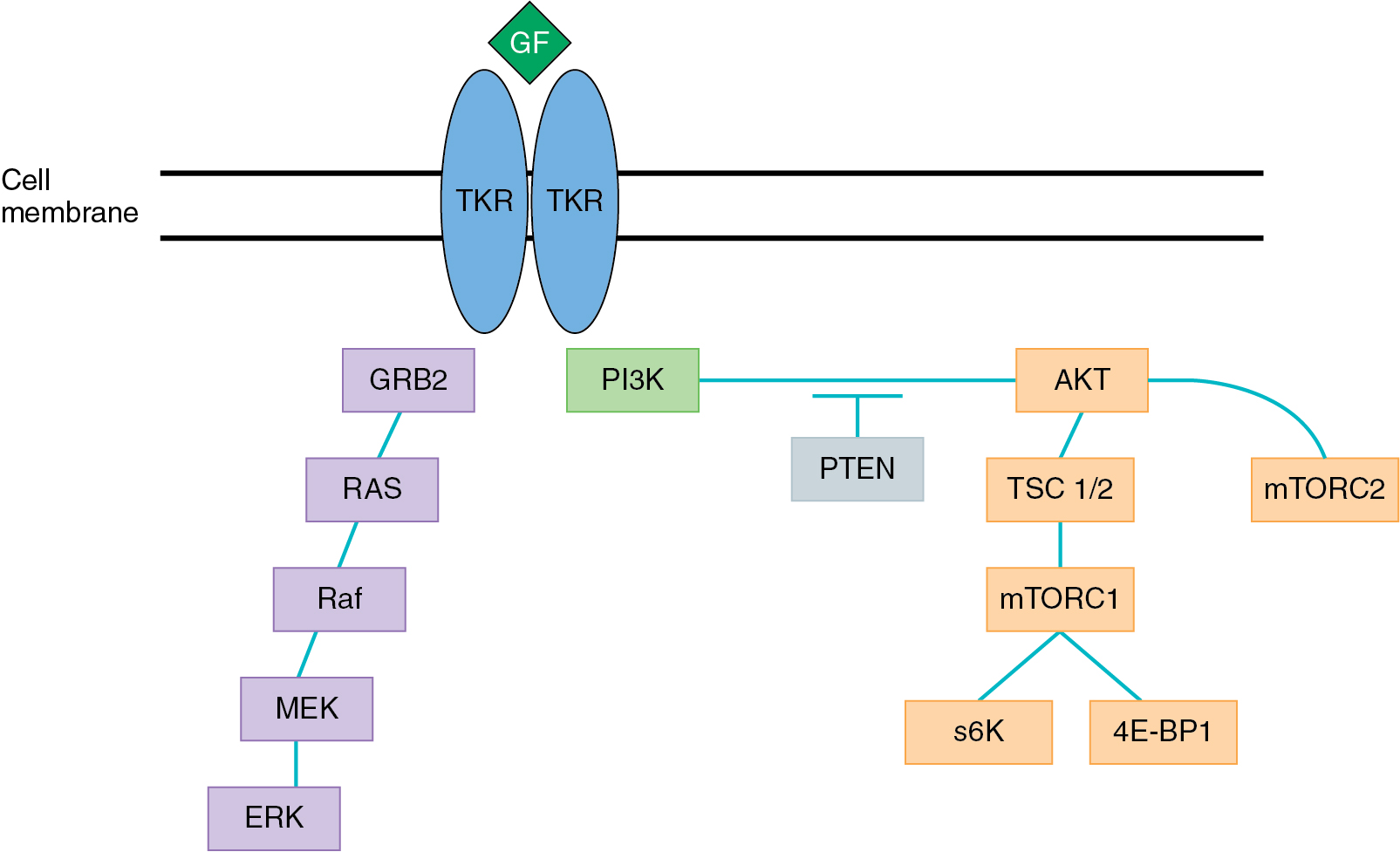
The PI3K/AKT pathway has been shown to be activated in a variety of cancers including gynecologic malignancies. The phosphatase tensin homolog on chromosome 10 (PTEN), an antagonist of PI3K signaling, is a tumor suppressor gene that converts PIP3 back to PIP2. Mutations in this gene are commonly encountered in gynecologic malignancies. Changes that result in loss of function leads to an overaccumulation of PIP3, causing unopposed signaling via the AKT pathway. Various other genetic alterations cause increased activation of the PI3K/AKT pathway and have been seen in both ovarian and endometrial malignancies ( Table 16.4 ). Therefore, targeting PI3K/AKT signaling has become a strategy of interest in the treatment of gynecologic cancers. mTOR is a key protein in the PI3K/AKT pathway, consisting of two major complexes – mTORC1 and mTORC2. Agents targeting mTOR compete with ATP at the catalytic site, blocking activity of both mTORC1 and mTORC2. To date, however, there are no approved indications for inhibitors of the PI3K/AKT/mTOR pathway for gynecologic malignancies.
| Ovarian Cancer | Endometrial Cancer | Reference | |
|---|---|---|---|
| PTEN | 5.5% | 45% | 84 |
| PIK3CA | 6% | 39% | 85 |
| AKT1 | 2% | 4% | 86 |
| AKT2 | 13.3% | 3.1% | 87 |
Poly-adenosine diphosphate-ribose polymerase pathway
As previously discussed, DNA repair mechanisms represent critical cellular processes that maintain high fidelity DNA replication. DNA repair is frequently altered in tumor cells, promoting the propagation of genomic mutations and ultimately aberrant cellular growth. Uncontrolled cellular division is a hallmark of cancer development, and sufficient DNA repair activity is needed to sustain this level of growth. PARP is a protein involved in repair of single-stranded DNA breaks via the base excision repair pathway. When PARP activity is inhibited, this leads to a limited ability to repair single-stranded breaks which ultimately leads to double-stranded breaks which are lethal. This process is exploitable in tumor cells, especially those that harbor BRCA mutations. Cells that are deficient in BRCA activity are unable to repair double-stranded DNA damage. By targeting PARP activity in BRCA mutated tumor cells, this blocks single-stranded DNA repair resulting in unrepairable double-stranded breaks which leads to cell death. This concept is referred to as synthetic lethality. While BRCA mutated cells represent a specific class of tumors susceptible to PARP inhibition via synthetic lethality, this principle is applicable to all cells with deficient homologous repair activity. Ovarian cancer is a specific tumor type that is linked to inheritable genetic mutations. Approximately 20% of ovarian cancer patients have a BRCA mutation, and 50% have some genomic aberration that confers homologous repair deficiency. This has led to intense investigation of PARP inhibitors in ovarian cancer patients to exploit the errors in these repair mechanisms. The use of PARP inhibitors was initially studied in the recurrent setting, specifically demonstrating benefit in patients with platinum-sensitive disease. However, there has been a recent series of clinical trials establishing the role of PARP inhibitors in the upfront setting resulting in the US Food and Drug Administration (FDA) approval of PARP inhibitors in both the upfront and recurrent setting ( Table 16.5 ).
| Drug | Clinical Trial | Indications |
|---|---|---|
| Olaparib | Study 42 | Monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer who have been treated with ≥3 prior lines of chemotherapy. |
| Solo-2 Study 19 | Maintenance treatment in patients with recurrent epithelial ovarian cancer who had CR or PR to platinum-based chemotherapy. | |
| Solo 1 | Maintenance therapy in patients with epithelial ovarian cancer who had CR or PR to platinum-based chemotherapy and a deleterious BRCA mutation (germline or somatic). | |
| PAOLA | Maintenance therapy in combination with bevacizumab in patients with advanced epithelial ovarian cancer who had a CR or PR following first-line platinum-based chemotherapy | |
| Rucaparib | Study 10 ARIEL2 | Monotherapy for treatment of patients with deleterious BRCA mutation (germline or somatic) ovarian cancer following ≥2 lines of chemotherapies. |
| ARIEL3 | Maintenance therapy in patients with recurrent epithelial ovarian cancer who had CR or PR to platinum-based chemotherapy. | |
| Niraparib | NOVA | Maintenance therapy in patients with recurrent epithelial ovarian cancer who had CR or PR to platinum-based chemotherapy. |
| QUADRA | Monotherapy in patients with recurrent epithelial ovarian cancer following ≥3 lines of chemotherapy with tumor positive for HR deficiency. | |
| PRIMA | Maintenance therapy in patients with advanced epithelial ovarian cancer who had a CR or PR following first-line platinum-based chemotherapy. |
Olaparib
Olaparib was the first PARP inhibitor studied in clinical trials. After promising results in early phase I trials, Study 19 was initiated as an international, randomized, double-blind, placebo-controlled, phase II trial investigating the use of Olaparib maintenance therapy in platinum-sensitive ovarian cancer patients. This study met its primary endpoint of improvement in progression-free survival (PFS) with median PFS of 8.4 months compared to 4.8 months (hazard ratio [HR] 0.35; P < .001). In a pre-planned analysis, patients with BRCA mutations received the biggest benefit (PFS of 11.2 months vs. 4.3 months). SOLO2 was a phase III trial investigating Olaparib as monotherapy in patients with recurrent ovarian cancer. Patients in this trial had a germline BRCA mutation and were randomized to receiving Olaparib or placebo as maintenance therapy following at least two lines of prior chemotherapy. Treatment with maintenance therapy Olaparib resulted in a significantly increased PFS (19.1 vs. 5.5; HR: 0.30; P < .0001). These trials led to an FDA approval of Olaparib as maintenance therapy in recurrent ovarian cancer for patients who had a complete response (CR) or partial response (PR) to their most recent line of platinum-based chemotherapy.
Recent data has been published demonstrating the benefit of Olaparib in the upfront setting. The SOLO-1 trial studied the efficacy of Olaparib maintenance in patients with a germline or somatic BRCA mutation following response to upfront platinum-based chemotherapy. At the time of publication, results from the trial showed a drastic improvement in patients treated with Olaparib maintenance therapy compared to placebo. While median PFS had not yet been reached at the time of publication, estimated improvement was 36 months compared to placebo, with a 70% reduction in risk of progression or death. These unprecedented results led to FDA approval of Olaparib as maintenance therapy in BRCA mutated patients with ovarian cancer who had a CR or PR following upfront platinum-based chemotherapy. As previously discussed, the phase III trial PAOLA-1 (Platine, Avastin, and Olaparib in 1st Line) studied the addition of Olaparib and bevacizumab maintenance therapy compared to bevacizumab alone following completion of chemotherapy. In the intention to treat (ITT) analysis, PFS improved by almost 6 months (22.1 vs. 16.6 months; HR: 0.59; P < .0001). The largest magnitude of benefit was found in patients with homologous recombination deficiency (HRD) including BRCA mutation (PFS 37.2 vs. 17.7 months; HR: 0.33). This has led to an additional approval for Olaparib in combination with bevacizumab in ovarian cancer patients with HRD as maintenance therapy following a CR or PR to upfront chemotherapy.
Rucaparib
Rucaparib is a potent inhibitor of both PARP-1 and PARP-2. Study 10 was a three-part phase I/II trial evaluating the efficacy of rucaparib in patients with recurrent ovarian cancer. In part one, the recommended phase II dose (RP2D) was established at 600 mg BID. This dose was applied in part 2A in the phase II portion of the trial. This dose was given to 42 patients with platinum-sensitive HGSOC carrying a germline BRCA mutation. Objective response rate for these patients was 59.5% with a median duration of response of 7.8 months. In part 2B, the patient population studied had a germline or somatic BRCA mutation and had received two to four lines of prior chemotherapy. Finally, part 3 was designed to study the pharmacokinetics of higher doses in any solid tumors, including ovarian cancer with BRCA mutations. Results of part 2B and part 3 are still pending, although recruitment has been completed. Similarly, rucaparib was evaluated in a collection of trials in the Assessment of Rucaparib in Ovarian Cancer Trial (ARIEL) series. ARIEL 2 is an ongoing two-part phase II open label trial. In part 1, patients enrolled with recurrent HGSOC or endometrioid ovarian cancer after treatment of one or more line of chemotherapy. Close to 200 patients with platinum-sensitive recurrent ovarian cancer were enrolled and stratified by BRCA and HRD status. The study used a LOH score as a biomarker for HRD status. Patients with a BRCA mutation experienced a PFS of 12.8 months with rucaparib compared to 5.2 months in the comparator group of BRCA wild type and LOH low patients. There was also a small but significant improvement in PFS for patients with wild type BRCA and LOH high (5.7 months vs. 5.2 months; HR: 0.62; P = .011). Similarly, patients with a BRCA mutation had an 80% overall response rate (ORR) and 29% in BRCAwt/LOH high compared to 10% in BRCAwt/LOH low patients. This trial showed the benefit of rucaparib in the platinum-sensitive recurrent setting, but also demonstrated the validity of using LOH as a marker of HR deficiency. Part 2 of ARIEL 2 is currently ongoing. In a pooled analysis from Study 10 and ARIEL 2, the benefit of rucaparib use was confirmed. Patients included in the analysis had HGSOC and a deleterious BRCA mutation who received at least two prior lines of chemotherapy. ORRs were 53.8% with 8.5% achieving CR and 45.3% experiencing a PR. ARIEL 3 is a phase III placebo-controlled trial evaluating maintenance rucaparib in patients with platinum-sensitive disease following response to their most recent course of platinum-based chemotherapy. This trial had three nested cohorts studied: (1) patients with germline or somatic BRCA mutations, (2) HRD including patients with BRCA mutations and LOH high, and (3) the intent to treat population. Maintenance rucaparib significantly improved PFS in all three cohorts with acceptable toxicity. The results from Study 10 and ARIEL trials led to two FDA-approved indications for rucaparib in patients with ovarian cancer: (1) as monotherapy in patients with recurrent ovarian cancer and a germline or somatic BRCA mutation after at least two lines of prior chemotherapy, and (2) maintenance therapy in platinum-sensitive recurrent ovarian cancer after a CR or PR to most recent platinum-based chemotherapy.
Niraparib
Niraparib was first studied in phase I trials and identified the maximum tolerated dose of 300 mg daily with an ORR of 40% in patients with recurrent ovarian cancer. This led to evaluation in the double-blind randomized controlled phase III NOVA trial. As in previous studies, patients with platinum-sensitive recurrent ovarian cancer were randomized to niraparib maintenance versus placebo. The two cohorts analyzed in this trial were patients with and without deleterious BRCA mutations. Additional subgroup analyses were performed to study the effect in patients with HRD with and without BRCA mutations as well as BRCA wild-type patients. Once again, PARP inhibition use showed benefit across all cohorts. In the predetermined groups, PFS was 21.5 months with niraparib use compared to 5.5 months in the placebo arm in patients with BRCA mutation. In patients without a BRCA mutation, PFS improved by 5.4 months with niraparib (9.3 vs. 3.9 months; HR: 0.45; P < .001). Just as with Olaparib and rucaparib, results from NOVA led to FDA approval for niraparib as maintenance therapy in patients with recurrent platinum-sensitive ovarian cancer. Recent data was published from QUADRA, an open-label, single-arm, phase II trial which evaluated niraparib in heavily pre-treated patients with recurrent ovarian cancer. Patients were treated with three or more previous lines of chemotherapy. The primary efficacy group was patients who were HRD and platinum-sensitive with three or four lines of prior therapy. In this group, 27.5% of patients achieved an objective response and a 68.6% disease control rate. This trial demonstrated the benefit of niraparib in this heavily pre-treated group where effective treatment options are limited. Niraparib has been approved for monotherapy in patients with recurrent ovarian cancer and HRD following at least three lines of prior chemotherapy.
PRIMA is a recently completed trial investigating the use of niraparib maintenance in the upfront setting in patients with newly diagnosed ovarian cancer. Patients included had advanced HGSOC or endometrioid ovarian carcinoma with a CR or PR to first-line platinum-based chemotherapy. Patients underwent HRD testing prior to randomization and were randomized 2:1 to niraparib maintenance versus placebo. Stratification factors in this trial included PR versus CR response to primary chemotherapy, the use of neoadjuvant chemotherapy, and HRD status. The primary endpoint of the study was PFS. In the intention to treat group, PFS improved by 5.6 months with niraparib maintenance (13.8 vs. 8.2 months; HR: 0.62; P < .001). The magnitude of benefit was increased to 11.5 months in patients who were homologous recombination deficient (21.9 vs. 10.4 months; HR: 0.43; P < .001). The benefit was not as profound in the subgroup of patients who were homologous recombination proficient. In this group niraparib improved PFS by 3 months (8.1 vs. 5.4 months; HR 0.68). Quality of life was maintained in the experimental arm and no new safety signals were observed. Niraparib maintenance has now been FDA-approved for all patients with endometrioid or HGSOC with a demonstrated response to upfront chemotherapy regardless of HRD status.
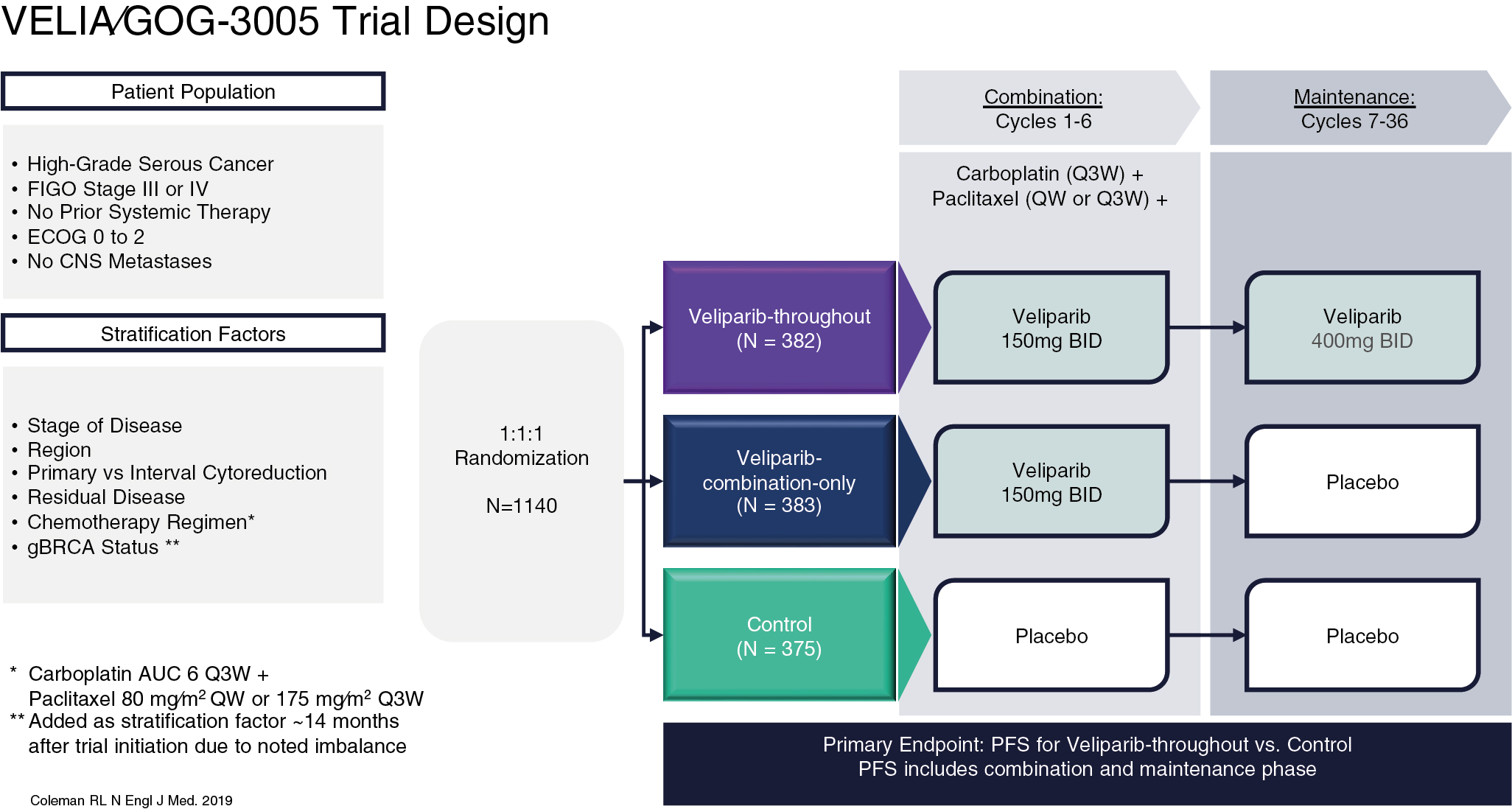
Veliparib
Veliparib is an inhibitor of PARP-1 and PARP-2 but demonstrates lower levels of PARP trapping activity compared to other previously described PARP inhibitors in this chapter, theoretically reducing the potency of the drug. Phase II data showed that efficacy of veliparib in patients with BRCA mutations demonstrated a 26% response rate. However, the unique advantage of veliparib is the ability to combine the PARP inhibitor with systemic chemotherapy with an acceptable side effect profile, which is perhaps reflective of its weaker activity. This led to the VELIA trial, an international, phase III, randomized placebo-controlled trial. In this study, patients with newly diagnosed advanced ovarian cancer were randomized to one of three arms ( Fig. 16.6 ). The control group were treated with primary carboplatin and paclitaxel with concurrent placebo therapy followed by placebo maintenance. In the combination only arm, veliparib was administered concurrently with upfront carboplatin and paclitaxel and given maintenance placebo therapy. Finally, in the veliparib throughout arm, the patients were treated with concurrent chemotherapy and veliparib followed by veliparib maintenance. The primary endpoint evaluated PFS against the placebo-control arm. The use of veliparib throughout resulted in a 6.2-month benefit in PFS compared to the control group (23.5 vs. 17.3 months; HR: 0.68; P < .001). In sub-group analyses there is a similar amplification of benefit in PFS for patients with BRCA mutations (BRCAm) specifically, as well as those with HRD. PFS in BRCAm patients was 34.7 months compared to 22.0 months (HR: 0.44; P < .001) and was 31.9 months in HRD patients compared to 20.5 months (HR: 0.57; P < .001). Adverse events were consistent with chemotherapy during the combination and maintenance phase of the experimental veliparib throughout arm. A similar number of chemotherapy cycles were completed when patients were given concurrent veliparib therapy, demonstrating a safety profile that did not impact the ability of patients to received standard of care chemotherapy.