Fig. 5.1
Landscape of genetic variation in high-risk neuroblastoma (Nature Genetics 2013). MYCN amplification, deletion of chromosomes 1p, 3p, and 11q, gain of chromosomes 1q, 2p, and 17q, as well as genetic alterations of ALK, ATRX, and other genes are depicted in the landscape [9] This figure is from a paper written by Trevor J Pugh et al. entitled “The genetic landscape of high-risk neuroblastoma,” which was published in Nature Genetics in 2013
5.2 Chromosomal Abnormalities
Low-stage neuroblastoma is marked by numeric changes of chromosomal copy number, whereas high-stage neuroblastoma is marked by segmental chromosomal aberrations (gains or losses of chromosomal fragments) that result in hemizygous deletion of the chromosome arms 1p and 11q and gain of chromosome 17q. These tumor-associated segmental chromosome aberrations, MYCN amplification and DNA diploidy, are associated with poor prognosis for these neuroblastoma patients [1, 3, 17, 18]. Moreover, recent next-generation sequencing of neuroblastoma has identified a new chromosome alteration phenomenon termed chromothripsis [12].
5.2.1 MYCN Amplification
MYCN oncogene amplification on chromosome 2p24 is associated with advanced stage disease and poor outcome and occurs in approximately 20 % of primary neuroblastoma tumors and approximately 40 % of high-risk neuroblastomas [3, 17, 19–21]. MYCN amplification has been used for treatment stratification of neuroblastoma patients since the late 1980s [1]. In addition to the chromosome 2p locus, MYCN amplification also maps to the cytogenetic manifestations of gene amplification: the double-minute chromatin bodies (DMs) or homogenously staining region (HSRs). DMs may accumulate by uneven segregation during mitosis, and HSR may arise by integration of the amplified DNA into different chromosomal loci [22, 23]. Neuroblastoma tumors with MYCN amplification usually express MYCN at higher levels than tumors without amplification. It has been reported that MYCN expression is inversely correlated with survival probability [24], but this correlation may only be limited to stage 2, 3, and 4 neuroblastomas, the latter which contains the high-risk group of patients [25, 26]. MYCN genomic amplification with overexpression serves as prognostic markers for survival. Stage 4 patients with MYCN amplification but without overexpression had no increased likelihood of death, whereas cases with MYCN overexpression but no genomic amplification showed low survival [27]. Recent transcriptome analyses have identified a MYC/MYCN gene signature that identifies patients with a poorer prognosis even in the absence of MYCN amplification [28]. MYCN was first identified as a bona fide oncogene with the development of the tyrosine hydroxylase-MYCN (TH–MYCN) transgenic mouse model in which mice transgenic for tyrosine hydroxylase promoter-driven MYCN expression develop tumors similar to human neuroblastomas [29]. This was supported by studies in which knockdown of MYCN in neuroblastoma cell lines suppressed cell proliferation [30, 31]. The MYCN gene encodes N-Myc, a helix-loop-helix/leucine zipper transcription factor that regulates genes involved in cell cycle progression, apoptosis, and stemness. Genes regulated by MYCN include the self-renewal protein BMI1, anaplastic lymphoma kinase (ALK), tumor suppressor p53, antiproliferative protein Dickkopf-1, histone chaperone FACT (facilitates chromatin transcription), and myriad polyamine enzyme ODC1 [32–37]. N-Myc dimerizes with MAX to activate gene transcription and forms a repression complex with transcription factor SP1 and MIZ1 to repress gene transcription [38, 39]. N-Myc has also been reported to associate with EZH2, a subunit of polycomb repressor complex 2 to repress tumor suppressor gene clusterin, suggesting a role for N-Myc in epigenetic regulation [40, 41]. Moreover, although not formally proven, it is possible that MYCN, like MYC, may function as a universal amplifier of gene transcription [42, 43]. Knockdown of MYCN decreases cell proliferation, and overexpression of MYCN suppresses neuroblastoma cell differentiation and promotes proliferation and cell cycle progression presumably via regulation of its target genes. However, the critical molecular mechanisms and signaling pathways by which MYCN stimulates tumorigenesis remain to be critically investigated.
5.2.2 TERT Rearrangement
Whole-genome sequencing (WGS) of 56 neuroblastomas identified recurrent genomic rearrangements proximal to the telomerase reverse transcriptase gene (TERT) locus (chromosome 5p15.33). These events occurred predominantly in high-risk neuroblastoma (12 of 39 high-risk neuroblastomas) in a mutually exclusive fashion with MYCN amplifications and ATRX mutations [44]. TERT rearrangements were validated in 217 additional cases, and TERT rearrangements define a subgroup of high-risk neuroblastomas with poor outcome [44]. In a similar whole-genome sequencing study, structural rearrangements of TERT were identified in 17 of 75 high-stage neuroblastomas (23 %). Consistent with previous studies, TERT rearrangements, ATRX deletions, and MYCN amplifications were detected in high-risk neuroblastomas, and they were mutually exclusive and associated with poor prognosis [45]. From these studies, TERT rearrangements were identified to be the second most frequent genetic alteration after MYCN amplification [44, 45]. Induced transcriptional upregulation of TERT is caused by the acquisition of a super enhancer or strong enhancer by diverse structural genomic rearrangements [44, 45]. In normal cells, telomeres shorten with every cell cycle to restrict lifespan. However, in cancer cells, the telomeres can be preserved through alternative lengthening of telomeres (ALT) or activation of TERT. TERT can be activated by mutations of the promoter region or upstream rearrangements [46, 47]. In neuroblastoma, no mutations have been identified in the TERT promoter region (10). Instead, structural rearrangements at the TERT locus lead to activation of its transcription [44, 45]. Furthermore, in MYCN amplified neuroblastomas without TERT rearrangements, MYCN functions as a transcription activator of TERT leading to elevated levels of TERT mRNA although not to the extent seen in tumors with TERT rearrangements [44, 48]. Importantly, the majority of the high-risk neuroblastomas are affected by either MYCN amplification, TERT activation, or ATRX mutations, all of which lead to telomere lengthening and contribute to the immortality of this subtype of neuroblastomas [44, 45].
5.2.3 Alterations of Chromosome
Even though MYCN status is a prognostic biomarker in neuroblastoma, segmental chromosomal aberrations (gains or losses of chromosomal fragments) can also be used to assess neuroblastoma aggressiveness. Loss of chromosomes 1p, 3p, 4p, 14q, 16p, and 19q or gain of chromosomes 1q, 2p, 7q, 9p, 11p, 12q, and 17q are observed in neuroblastoma [3, 49, 50]. Among them, deletion of chromosomes 1p and 11q and gain of 17q are the most frequently occurring chromosomal alterations in high-stage disease [1, 17, 18, 51, 52]. A study from the international neuroblastoma risk group (INRG) analyzing a cohort of 8,800 children diagnosed with neuroblastoma shows that deletions of 1p or 11q and gain of 17q are significantly associated with poor outcome of patients [17].
5.2.3.1 Alterations of Chromosome 1p
Loss of heterozygosity (LOH) at chromosome 1p occurs in 30–35 % of primary neuroblastomas. In addition, 1p LOH is frequently found in unfavorable neuroblastoma and is associated with MYCN amplification [3, 17, 53, 54]. LOH at chromosome band 1p36 is the smallest region of overlap, and 1p36 LOH is independently associated with a poor outcome in patients with neuroblastoma [55]. The frequent LOH on chromosome 1p raises the possibility that this region contains tumor suppressor genes and the loss of such tumor suppressor genes contributes to tumorigenesis. Consistent with this hypothesis, reintroduction of chromosome 1p into neuroblastoma induces differentiation and cell death [56]. Recent studies have shown that multiple genes on chromosome 1p suppress neuroblastoma cell growth; these candidate neuroblastoma tumor suppressor genes include CHD5, CASZ1, CAMTA1, KIF1Bß, and microRNA-34a [57]. CHD5 is the commonly deleted tumor suppressor genes in neuroblastoma. It is a member of the chromodomain superfamily, which is a key component of nucleosome-remodeling complexes (NuRD) [58]. Expression of CHD5 is silenced through both 1pLOH and promoter methylation [59]. High CHD5 expression is strongly associated with favorable event-free and overall survival even after correction for MYCN amplification and 1p deletion [60]. Forced expression of CHD5 in neuroblastoma cells with low endogenous CHD5 suppresses neuroblastoma growth in mouse xenografts [60]. CASZ1 encodes a zinc finger transcriptional factor, and low expression of CASZ1 is associated with poor prognosis of neuroblastoma [61, 62]. Biallelic inactivation of CASZ1 by both 1pLOH and EZH2-mediated epigenetic silencing leads to its decreased expression in neuroblastomas of poor outcome [63]. CASZ1 transcriptional activity is mediated by recruitment of NuRD complexes, and the recent identification of binding between CASZ1 and CHD5 indicates a potential interaction between these two neuroblastoma tumor suppressors [64]. Restoration of CASZ1 in neuroblastoma cells induces cell differentiation, inhibits cell proliferation and migration, and suppresses neuroblastoma xenograft growth [61]. The expression of CAMTA1 (calmodulin-binding transcription activator 1) is an independent predictor of poor outcome of neuroblastoma and inhibits neuroblastoma cell growth and activates differentiation program [65]. KIF1Bß is a member of the kinesin superfamily proteins, and hemizygous deletion of KIF1Bß in primary neuroblastoma is correlated with advanced stages and MYCN amplification [66]. Forced expression of KIF1Bß in neuroblastoma cells results in apoptosis [66, 67]. Micro RNA miR–34a is under-expressed in unfavorable neuroblastomas and reintroduction of miR-34a in neuroblastoma cell lines with low endogenous miR–34a suppressed cell proliferation [68, 69]. Until now, no recurrent nonsynonymous mutations have been identified in these candidate 1p36 tumor suppressor genes. Moreover, the link between the loss of any of these candidate 1p tumor suppressors and the development of neuroblastoma tumors in mice has not been reported. It is possible that the haploinsufficiency of multiple genes at the 1p locus is required for neuroblastoma tumorigenesis.
5.2.3.2 Alterations of Chromosome 11q
Allelic loss of chromosome 11q occurs in approximately 33–43 % of neuroblastomas patients and is associated with poor prognosis. Importantly, deletion of 11q is inversely associated with 1p deletion and MYCN amplification (Fig. 5.1), which makes it a potential biomarker for the aggressive tumors in patients without MYCN amplification [4, 55, 70, 71]. Within the chromosome bands 11q22-q23, the region that is most frequently lost in neuroblastoma [70–72], there are several candidate tumor suppressor genes including ATM, TSLC1, and CADM1. ATM is the gene mutated in ataxia telangiectasia (AT), and knockdown of ATM in neuroblastoma cell lines with intact 11q promotes neuroblastoma progression in soft agar assays and in subcutaneous xenografts in nude mice [73]; TSLC1, a tumor suppressor in lung cancer 1, suppresses neuroblastoma cell proliferation [74]; CADM1, cell adhesion molecule 1, when overexpressed in neuroblastoma cells, results in a significant reduction in colony formation on soft agar [75, 76]. ATM is involved in the DNA repair pathway, and alteration in its activity is consistent with the finding that high-risk neuroblastoma tumors with the 11q loss display a chromosome instability phenotype [73]. Like 1p deletion, no recurrent inactivating mutations have been found in these genes at 11q; thus, whether these genes are bona fide tumor suppressor genes need to be further investigated.
5.2.3.3 Alterations of Chromosome 17q
Gain of distal chromosome arm 17q (17q21-qter) is the most common aberration found in neuroblastomas and occurs in 50–70 % of tumors. Gain of 17q is frequently associated with 1p LOH, 11q LOH, MYCN amplification, older age, and advanced stage [77–79]. Interestingly, in the neuroblastoma tumors that developed from MYCN transgenic mice, the murine equivalent of chromosome 17 is the most commonly gained chromosome [29]. Consistently, overexpression of MYCN in neuroblastoma cell lines with a single copy of MYCN and intact 17q leads to an unbalanced gain of 17q [29, 80]. These results suggest a functional relationship between MYCN overexpression and the gain of 17q in neuroblastoma. Although the mechanism by which 17q gain results in a more aggressive malignancy is not clear, the gain of 17q does lead to increased expression of the 17q resident anti-apoptotic protein BIRC5/survivin [81, 82], which might contribute to neuroblastoma progression.
5.2.4 Chromothripsis
At the level of genome structure, a local shredding of chromosomes with subsequent random reassembly of the fragments (localized massive genome rearrangements) has been found in some types of cancers. This phenomenon has been termed chromothripsis [83, 84]. Whole-genome sequencing (WGS) in 87 untreated primary neuroblastoma tumors of all stages identified a number of genetic alterations, and an analysis of the frequency of the structural variations per chromosome showed that chromothripsis occurred in ten tumors [12]. Chromothripsis was not found in low-stage tumors but in 18 % of the stage 3 and 4 neuroblastoma, although the prognostic impact of chromothripsis was not independent of age and stage in a multivariate analysis. Chromothripsis was associated with MYCN amplification, 1p LOH, and a poor prognosis. In one tumor from this study, chromothripsis resulted in MYC (c–Myc) amplification and high expression [12]. In another WGS study, chromothripsis occurred at the TERT locus in 5 of 75 high-stage neuroblastomas, leading to structural rearrangements and high expression of TERT gene [45]. Chromothripsis-induced structural aberrations frequently affected oncogene expression, which might contribute to neuroblastoma tumorigenesis. The finding that an inactivating deletion in FANCM occurred in one chromothripsis tumor and missense mutation in FAN1 in another tumor suggests that inactivating events in the fanconi anemia signal transduction and DNA repair pathway in neuroblastoma may be associated with chromothripsis [12].
5.3 Genetic Mutations
Cancer is a genetic disease, and cancer develops when genetic mutations occur in genes that affect cell proliferation and differentiation. Familial neuroblastoma, in which mutations in neuroblastoma-associated genes are inherited, is observed in 1–2 % of patients [85]. In contrast, sporadic neuroblastoma occurs in most patients and is typified by nonheritable somatic mutations. Recent studies using next-generation sequencing techniques discovered germline mutations and recurrent somatic mutations in primary neuroblastoma, but these cases are rare. Almost 90 % of familial neuroblastomas contain ALK mutations with the remaining predominantly having mutations in PHOX2B. In sporadic neuroblastomas, ALK, ATRX, PTPN11, MYCN, NRAS, ARID1A, and ARID1B are the major genes in which somatic mutations occur, but their frequency is low, from 1.7 to 11 % [9–12]. Other rare cases having recurrent somatic mutations in primary neuroblastoma include protein tyrosine phosphatase, receptor type D (PTPRD), odd Oz/ten-m homolog 3 (ODZ3) and T-cell lymphoma invasion and metastasis 1 (TIAM1) [9, 10, 12]. This is consistent with the observation that pediatric malignancies have a lower frequency of somatic mutation than most adult cancers [86].
5.3.1 Germline Mutations
5.3.1.1 ALK
Next-generation sequencing of primary neuroblastoma samples elucidated activating mutations in anaplastic lymphoma kinase (ALK) in most hereditary and 7–8 % of sporadic neuroblastomas [87–91]. The ALK gene maps chromosome 2p23 and encodes a receptor tyrosine kinase (RTK). ALK is expressed in the central and peripheral nervous system, and in drosophila, ALK plays a critical role during the nervous system development [92–94]. The oncogenic activity of ALK was identified when it was found to be fused to nucleophosmin (NPM1) through a 2;5 chromosome translocation in most anaplastic large cell lymphomas (ALCL). Thus far, over 20 different genes have been described as being translocated with ALK, and ALK fusions have been found in diffuse large B-cell lymphomas, inflammatory myofibroblastic tumors, esophageal squamous cell carcinomas, and non-small-cell lung carcinomas [93, 95, 96]. ALK and ALK fusion proteins regulate cell survival, proliferation, and differentiation through the activation of JAK–STAT, PI3K–AKT, mTOR, sonic hedgehog (SHH), or CRKL–C3G–RAP1 GTPase pathways in different cell types [96, 97]. Unlike other cancers, no ALK fusions were observed in neuroblastoma; instead, point mutations and amplifications leading to aberrant ALK overexpression and activation were found in neuroblastoma [98]. Germline mutations discovered in neuroblastoma include R1275Q, R1192P, G1128A, T1151M, T1087I [87–90], and, in rare cases, the more potent F1245V and F1174V ALK mutations [99]. The R1275Q, R1245V, R1192P, F1174V, and T1151M mutations fall within the kinase activation domain. The G1128A mutation occurs at the third glycine of the glycine loop, and identical mutations of this glycine residue to alanine in BRAF have been shown to increase kinase activity [87, 89, 99]. Mutant ALK R1275Q occurs in many neuroblastoma cell lines, and cells that express mutant ALK R1275Q but not wild-type ALK exhibit strong kinase activity indicating constitutive activation [88]. Consistently, knockdown of ALK in neuroblastoma cells that express mutant ALK R1275Q resulted in a significant decrease in cell proliferation and colony formation on plastic [88], supporting an oncogenic role of ALK mutants in neuroblastoma. The incidence of ALK mutations is about 8 % in neuroblastoma, and they occur in all stages although the frequency of ALK alterations is higher in high-risk neuroblastoma, with 10 % containing point mutations and 4 % containing ALK amplification [87–91]. Somatic mutations of ALK will be described later in this review.
5.3.1.2 PHOX2B
A majority of patients with congenital malformation of neural crest-derived cells such as Hirschsprung disease (HSCR) and congenital central hypoventilation syndrome (CCHS) harbor paired-like homeobox 2b (PHOX2B) mutations, and neuroblastoma occurs in a subset of these patients [100, 101], suggesting PHOX2B may be associated with neuroblastoma tumorigenesis. DNA sequence analysis showed that PHOX2B germline mutations occur in families with neuroblastoma; thus, PHOX2B can be considered a bona fide neuroblastoma predisposition gene when mutated in the germline [102, 103]. The PHOX2B gene maps to chromosome 4p12 and encodes a homeobox transcription factor. PHOX2B directly regulates ALK transcription in neuroblastoma, which provides a link between these two mutated pathways in familial neuroblastoma [104]. PHOX2B is the master regulatory gene for both central and peripheral automatic nervous system development in mice [105]. In zebrafish, loss of PHOX2B impairs sympathetic neuronal differentiation [106] and suggests that perturbations in PHOX2B-regulated differentiation may contribute to neuroblastoma tumorigenesis. Heterozygous germline mutations that have been described in neuroblastoma include R100L, R141G, and frameshift mutations. PHOX2B R100 amino acid has been shown to bind DNA of target genes, and PHOX2B R100L may lose DNA-binding ability but retain the ability to dimerize with the wild-type PHOX2B or other cofactors. Thus, PHOX2B mutations may result in loss of function or dominant-negative activities [102, 103]. Consistently, forced overexpression of wild-type PHOX2B but not mutant PHOX2B suppresses neuroblastoma cell proliferation and synergizes with all-trans retinoic acid to promote differentiation [107]. Introduction of a patient-derived PHOX2B mutation into the mouse phox2b locus recapitulates the clinical features of CCHS but did not give rise to neuroblastoma [108], suggesting that PHOX2B alone might not be sufficient to drive neuroblastoma tumorigenesis. Unlike ALK mutations, PHOX2B mutations only account for approximately 6.4 % of familial neuroblastoma [107]. Some other genes (TP53, EZH2, SDHB, PTPN11, HRAS, and NF1) may be considered candidate neuroblastoma predisposition genes [4], but these cases are very rare. Until now, the cause of the remaining familial neuroblastomas that do not harbor ALK or PHOX2B mutations remains unresolved.
5.3.2 Recurrent Somatic Mutations
5.3.2.1 ALK
As described above, activating mutations in ALK occurred in 7–8 % of sporadic neuroblastomas [87–91]. Comprehensive analyses of ALK mutations across 1,596 diagnostic neuroblastoma samples showed that over 10 different residues are mutated in ALK and most mutations occurred within the kinase domain, and the most commonly mutated residues accounted for 85 % of mutations are F1174 (30 %), R1245 (12 %), and R1275 (43 %) [91]. Across the whole cohort, the presence of ALK mutation correlated with reduced event-free survival (EFS) and overall survival (OS) [91]. Oncogenic transformation occurred when NIH3T3 cells were transfected with certain ALK mutation constructs. In some neuroblastoma cell lines with certain ALK mutations, the kinase was constitutively activated, and knockdown of ALK mutants or treatment with ALK inhibitor crizotinib suppressed cell proliferation [87–91], which indicates that ALK is an oncogenic driver in neuroblastoma. More convincingly, the targeted expression of the most aggressive ALK mutant F1174L alone induces neuroblastoma in transgenic mice [109]. Moreover, coexpression of ALK mutant F1174L and MYCN accelerated tumor onset and increased tumor penetrance compared with MYCN alone suggests a cooperative effect between these two oncogenes [109–112]. Interestingly, MYCN directly regulates ALK transcription, and ALK stimulates the kinase ERK5 to promote the expression of MYCN in neuroblastoma [34, 113], which provides a connection between these two oncogenes in neuroblastoma. Although F1174L is the most frequent and aggressive mutation in sporadic neuroblastoma, it has been rarely observed in familial neuroblastoma suggesting that this mutation may not be tolerated in the germline.
5.3.2.2 ATRX
A WGS study of 40 neuroblastoma tumors and subsequent validation sequencing of 60 neuroblastoma tumors showed that mutations in alpha thalassemia/mental retardation syndrome X-linked gene (ATRX) occurred in about 10 % of neuroblastoma and are enriched in older patients [9, 11, 12]. Based on age, ATRX mutations were found in 44 % of tumors from patients in the adolescent and young adult group (≥12 years), in 17 % of tumors from children (18 months- < 12 years), and 0 % of tumors from infants (0− < 18 months) [11]. ATRX maps to the X chromosome and encodes a SWI/SNF chromatin-remodeling helicase, which plays a role in chromatin remodeling, nucleosome assembly, and telomere maintenance [11, 114]. ATRX mutations are associated with X-linked mental retardation (XLMR) and alpha thalassemia. ATRX mutations have also been identified in pheochromocytomas and paragangliomas (PCC/PGL) and pancreatic neuroendocrine tumors [115, 116]. ATRX mutations include missense, nonsense, frameshift, or in-frame deletion, and they are mutually exclusive of MYCN amplification [11]. Neuroblastoma samples with ATRX mutations had complete or mosaic loss of the nuclear ATRX protein and show longer telomeres [11]. Most of the neuroblastomas with ATRX mutations had evidence of alternative lengthening of telomere (ALT) pathway activation, and this was consistent with findings that loss of ATRX/DAXX is associated with ALT in different cell lines [11, 117]. ATRX mutations were associated with age at diagnosis in children and young adults with stage 4 neuroblastoma. A larger study is required to determine the prognostic impact of ATRX mutations on survival [11]. How ATRX mutations lead to neuroblastoma progression remains unclear.
5.3.2.3 ARID1A and ARID1B
Next-generation sequencing, genome-wide rearrangement analyses, and targeted analysis of specific genomic loci of 71 neuroblastoma patients identified mutations in chromatin-remodeling genes AT-rich interaction domain 1A (ARID1A) and AT-rich interaction domain 1B (ARID1B) in eight cases (11 %). The mutations include missense, nonsense, frameshift, or in-frame deletion, and the ARID1A and ARID1B mutations were associated with decreased survival of neuroblastoma patients [10]. Both ARID1A and ARID1B are subunits of the SWI/SNF transcriptional complex [118, 119]. Mutations of ARID1A and ARID1B have also been identified in other types of cancer such as ovarian, endometrial, hepatocellular, breast, and microsatellite unstable colorectal cancers [120–125]. ARID1A and ARID1B have emerged as tumor suppressor genes [119, 124, 126]; thus, mutations of these genes may drive neuroblastoma tumorigenesis. Little is known as to how ARID1A and ARID1B mutations affect neuroblastoma tumorigenesis.
5.3.2.4 RAS
In primary neuroblastomas, mutations in neuroblastoma RAS viral oncogene homolog (NRAS) or Harvey RAS viral oncogene homolog (HRAS) are rare [9, 10], although mutations in the RAS–MAPK pathway occur more frequently in relapsed neuroblastoma [127]. WGS studies identified clonally enriched somatic mutations predicted to activate the RAS–MAPK pathway in 78 % relapsed neuroblastoma tumors (18 out of 23 patients). Activating mutations in the RAS–MAPK pathway were also identified in 60 % of tested neuroblastoma cell lines of which the majority have been established from relapsed neuroblastoma patients [127]. Mutated genes associated with the RAS-MAPK pathway include ALK, NRAS, HRAS, Kirsten RAS viral oncogene homolog (KRAS), B–Raf proto-oncogene, serine/threonine kinase (BRAF), protein tyrosine phosphatase non-receptor type 11 (PTPN11), fibroblast growth factor receptor 1 (FGFR1), and neurofibromin 1 (NF1) [127]. Another study also discovered new recurrent mutations in HRAS and KRAS, as well as CHD5, DOCK8, and PTPN14 in relapsed neuroblastomas [128]. Consistently, the constitutive activation of RAS–MAPK pathway was observed in neuroblastoma cell lines with MAPK pathway mutations.
5.4 Single Nucleotide Polymorphisms
The paucity of recurrent somatic mutations in neuroblastoma suggests that a complex genetic phenotype might contribute to the penetrance of neuroblastoma in the population. Genome-wide association study (GWAS) of DNA samples from neuroblastoma patients using high-density single nucleotide polymorphism (SNP)-based microarray has identified SNPs associated with different clinical features of neuroblastoma; the identified genes with SNPs include BARD1, CASC15, DDX4, DUSP12, HACE1, HSD17B12, IL31RA, LIN28B, LMO1, NBPF23, and NEFL [16, 129–138]. Among these genes, the SNPs of DDX4, DUSP12, HSD17B12, and IL31RA are associated with low-risk neuroblastoma, whereas the SNPs of BARD1, CASC15, HACE1, LIN28B, and LMO1 are associated with aggressive, high-risk disease [4, 15, 16, 137]. Moreover, BARD1, LIN28B, and LMO1 have been shown to be important predisposition genes and oncogenic drivers in neuroblastoma [16, 129, 130, 132–134].
5.4.1 BARD1
GWAS of 397 high-risk European-American neuroblastoma cases and 2,043 controls identified significant association of six SNPs at 2q35 within the BRCA1-associated RING domain 1 (BARD1) locus, which were further confirmed in a second series of 189 high-risk neuroblastoma cases and 1,178 controls [16]. Common BARD1 SNPs affect risk of neuroblastoma in African Americans based on the GWAS studies focused on 390 African-American neuroblastoma patients compared with 2,500 ethnically matched controls [139]. Similar association of BARD1 SNPs was observed in an Italian population (370 cases and 809 controls) [137]. BARD1 is an important regulator and interactor of the tumor suppressor BRCA1, yet it can also function by itself as a tumor suppressor [140]. In neuroblastoma, the disease-associated variations correlate with increased expression of the BARD1ß, which is an oncogenically activated isoform of BARD1. Overexpression of BARD1ß in NIH3T3 fibroblasts induces neoplastic transformation, and BARD1ß is required for neuroblastoma cell survival in vitro through interacting and stabilizing Aurora kinase B [134]. These results indicate SNPs of BARD1 are a major genetic contributor to neuroblastoma risk, and BARD1ß is an oncogenic driver of high-risk neuroblastoma.
5.4.2 LIN28b
GWAS of 2,817 neuroblastoma cases and 7,473 controls identified a significant association of SNP (rs17065417) within protein lin-28 homolog B (LIN28B) locus [129]. LIN28B is an RNA-binding protein that represses the let–7 family of microRNAs, and it is highly expressed and functions as an oncogene in many types of cancers [141–143]. LIN28B expression is higher, and let–7 microRNA expression is lower in neuroblastoma cell lines homozygous for the rs17085417 risk allele compared to heterozygous cell lines [129]. High LIN28B expression is associated with worse overall survival in primary neuroblastoma tumors and is an independent risk factor for adverse outcome in neuroblastoma [129, 131]. Knockdown of LIN28B in neuroblastoma cells leads to growth inhibition [129]. Importantly, in a mouse model expression of LIN28B in the sympathoadrenal lineage results in the development of neuroblastoma [131]. Suppression of let–7 microRNAs by LIN28B resulted in elevated MYCN expression in neuroblastoma cell lines, and similarly in the neuroblastoma developed from the LIN2B transgenic mouse, let–7 microRNA level is low and MYCN level is high [131]. Vice versa, MYCN upregulates LIN28B expression either directly through binding LIN28B promoter or indirectly through miR–26a–5p and miR–26b–5p [144]. The cross-regulation between LIN28B and MYCN provides a connection between these two oncogenes in neuroblastoma. LIN28B regulates expression of the oncogene RAN and cooperates with RAN to activate Aurora kinase A to drive neuroblastoma oncogenesis [130], identifying another pathway by which high levels of LIN28B expression drive neuroblastoma tumorigenesis.
5.4.3 LMO1
GWAS on 2,251 neuroblastoma patients and 6,097 control subjects of European-Americans identified a significant association within LIM domain only 1 (LMO1) at 11p15.4 (rs110419), and the signal was enriched in the subset of most aggressive neuroblastoma patients [133]. Similarly, a significant association within LMO1 (rs204926) was identified in a Chinese population [145]. Moreover, genome-wide DNA copy number alterations in 701 primary tumors identified 12.4 % with aberrations at the LMO1 locus, and this event was associated with more advanced disease [133]. LMO1 encodes a cysteine-rich transcriptional regulator, which has been implicated in cancer [146–148]. LMO1 expression levels are higher in primary tumors and neuroblastoma cell lines with SNP risk alleles and somatic copy number gains. Knockdown of LMO1 in neuroblastoma cells with high endogenous LMO1 levels inhibited cell proliferation, whereas overexpression of LMO1 in neuroblastoma with low LMO1 levels increased proliferation. A recent study identified another highly associated SNP (rs2168101 G > T) within the first intron of LMO1 gene locus that occurs in a super enhancer [132]. The ancestral G allele that is associated with tumor formation resides in a conserved GATA transcription factor-binding motif, while the more recently evolved protective TATA allele is associated with decreased total LMO1 expression and does not bind GATA3 [132].
5.5 MicroRNA
MiRNAs (microRNAs) are short RNAs (9–22 nucleotides in length) that regulate more than 60 % of human gene expression by causing mRNA degradation or inhibiting protein translation [149]. MiRNA profiling studies in neuroblastoma identified miRNAs that are differentially expressed in favorable and unfavorable, MYCN non-amplified and MYCN amplified, low-risk and high-risk neuroblastomas [150–154]. A miRNA profiling study using over 500 neuroblastoma samples identified a 25-miRNA gene signature that can significantly discriminate patients with respect to progression-free and overall survival, and this 25 miRNA gene signature can be used for risk stratification in both archived and fresh neuroblastoma tumor samples [155]. In the past decades, many miRNAs have been linked to tumor suppressor or oncogenic roles [156]. Recently miRNAs with tumor suppressor roles include miR–337–3p and miR–584–5p that target matrix metalloproteinase 14 [157, 158], miR–362–5p which targets phosphatidylinositol 3-kinase-C2beta [159], miR–449a targeting CDK6, miR–542–3p targeting survivin [160], and miR–375 targeting MYCN [161]. Oncogenic miRNAs include miR–558 that targets heparanase [162] and miR–421 that targets the tumor suppressor menin [163]. Interestingly, unlike the traditional miRNAs that decrease target gene mRNA or protein levels, miR–558 stimulates heparanase transcription via activation of epigenetic histone active marks and increases in RNA polymerase II on the heparanase promoter in neuroblastoma cells [162]. Among those miRNAs studied in neuroblastoma, miR–34a and miR–17–92 are the most commonly reported and best-characterized miRNAs. MiR–34a localizes to chromosome 1p36 and is frequently included in 1pLOH. MiR-34a is under-expressed in unfavorable neuroblastomas, and its reintroduction suppresses cell proliferation likely through targeting oncogenes E2F3 and MYCN and anti-apoptotic gene BCL2 [68, 69, 164]. The miR–17–92 cluster is directly activated by MYCN and plays an oncogenic role by targeting an apoptosis facilitator BIM, cell cycle inhibitor CDKN1A, as well as genes with tumor suppressor activity including TGF–beta and DKK1. Inhibition of miR–17–92 upregulates BIM and CDKN1A and suppresses tumor growth [165–167].
5.6 Long Noncoding RNA (lncRNA)
LncRNAs are ubiquitously expressed RNA molecules longer than 200 bp in length that lack protein-coding features and account for approximately 68 % of the human transcriptome of expressed genes [168]. LncRNAs have been recognized as integral components of the eukaryotic regulatory gene network [168, 169]. LncRNAs can act as a scaffold for the recruitment of epigenetic regulators such as polycomb repressive complex 2 (PRC2) and chromatin remodeling complex SWI/SNF to regulate gene transcription or serve as sponges to sequester miRNAs or proteins [168–172]. Almost 500 lncRNAs show 100 % conservation between human, rat, and mouse genomes, and these lncRNAs are defined as transcribed ultra-conserved regions (T-UCRs). The precise roles of these T-UCRs are unknown [173]. Real-time PCR analysis of the 481 T-UCRs in 34 high-risk, stage 4 neuroblastoma patients defined a 15 T-UCRs signature can be used to discriminate short- from long-term survivors in this group of patients [174]. The expression of a significant number of T-UCRs is altered after neuroblastoma cells are treated with all-trans retinoic acid (ATRA) [175]. T–UC.300A is downregulated by ATRA, and knockdown T–UC.300A in neuroblastoma decreased viability and invasiveness of ATRA-responsive cell lines [175], which suggests that some of the T-UCRs may play an oncogenic role in neuroblastoma. Very few recurrent mutations in protein-coding genes were identified in neuroblastoma [9–11]; thus, it highlights the importance of investigating nonprotein-coding RNA since approximately 70 % of the transcribed genome is transcribed into different types of RNAs other than protein-coding mRNAs [176, 177]. In fact, GWAS has shown that high-risk-associated SNPs are present in the introns of lncRNAs such as NBAT1 and CASC15 [129, 135]. Further studies showed that low-level expression of a short CASC15 isoform (CASC15–S) is associated with advanced neuroblastoma and poor patient survival, and CASC15–S regulates neuroblastoma cell proliferation and migration and functions as a tumor suppressor [178]. Similarly, the risk-associated lncRNA NBAT–1 controls neuroblastoma progression by regulating cell proliferation, migration, and neuronal differentiation and functions as a tumor suppressor [179]. LncUSMycN is a recently discovered lncRNA in neuroblastoma that is coamplified with MYCN in 88 of 341 human neuroblastoma tissues and is associated with poor patient prognoses [180]. Further study showed that lncUSMycN interacts with RNA-binding protein NonO, leading to N-Myc RNA upregulation and neuroblastoma proliferation. Furthermore, antisense oligonucleotides targeting lncUSMycN treatment suppressed neuroblastoma tumor growth in murine xenograft model [180]. In addition, many other lncRNAs were identified to play a role in neuroblastoma [181]. These findings suggest that lncRNAs may be implicated in neuroblastoma tumorigenesis.
5.7 Epigenetics
Cancer is driven by progressive genetic and epigenetic alterations [182]. Unlike adult cancer, fewer recurrent mutations have been identified in pediatric cancers including neuroblastoma [183]; thus, epigenetic alterations may be associated with certain subtypes of neuroblastomas. In fact, as described above, in the few recurrent genetic mutations present in neuroblastoma, ATRX, ARID1A, and ARID1B are chromatin regulators and key mediators of epigenetic regulation [10, 11]. Epigenetic regulation of gene expression is controlled by DNA methylation, modification of histones, and changes in nucleosome constitution, conformation, and structure.
Hypermethylation of CpG-island in gene promoters is a way to inactivate tumor-associated genes. Methyl-CpG-binding domain sequencing and methylation-specific PCR determined a 58-marker methylation signature that can be used to predict overall and event-free survival [184]. This is consistent with the other reports that the hypermethylation of some genes are associated with various clinical risk groups in neuroblastomas [185, 186]. In neuroblastoma, the most commonly reported genes that are silenced through promoter hypermethylation are RASSF1A, a tumor suppressor gene that interferes with RAS signaling [187], and CASP8, a tumor suppressor important in the death receptor-mediated apoptosis pathway [188]. Other methylated genes in neuroblastoma include CHD5, p14ARF, p16INK4a, CCND2, RARbeta2, TP73, RB1, DAPK1, HOXA9, TMS1, THBS1, MGMT, and TIMP–3 [59, 185, 189]. Importantly, many of these genes are involved in apoptotic signaling pathways; thus, DNA methylation-mediated silencing of these genes may contribute to the resistance or relapse of neuroblastoma tumors to cytotoxic chemotherapy.
Histone modifications such as acetylation, methylation, ubiquitination, and phosphorylation alter chromatin structure and affect the accessibility of transcription factors to DNA. In neuroblastoma, the majority of histone deacetylase (HDAC) family members are detectable, and some of them are aberrantly overexpressed [190]. Among these HDACs, high HDAC8 expression correlates with poor event-free and overall survival of neuroblastoma patients [191]. Knockdown of some of the individual HDAC family members in neuroblastoma cells leads to cell differentiation or apoptosis, and targeting HDACs with HDAC inhibitors suppresses tumor growth [191–193]. HDAC family members are candidate drug targets for neuroblastoma therapy.
The aberrant expression of other epigenetic modifiers that affect histone methylation and chromatin remodeling has also been observed in neuroblastoma: the lysine-specific demethylase, LSD1 (also named Lysine (K)-Specific Demethylase 1A, KDM1A), and the polycomb repressive complex1 (PRC1) subunit BMI–1 are overexpressed in poorly differentiated tumors, and silencing of LSD1 or BMI–1 induces differentiation [194, 195]. The 1p36 tumor suppressor gene CHD5 is a chromodomain protein that normally functions in nucleosome-remodeling complexes, and it is lost both through DNA methylation at the promoter region and 1p LOH. Restoration of CHD5 suppresses tumor growth [60]. As in other types of cancer, elevated levels of EZH2 are also found in undifferentiated neuroblastoma, and its methylation of H3K27 leads to the silencing of a number of genes in neuroblastoma with tumor suppressor activity such as CASZ1, NGFR, and CLU [63].
These studies indicate that there are alterations in the epigenome in neuroblastoma. Since enzymes that are potentially druggable regulate the epigenome, it provides a number of novel therapeutic options for neuroblastoma patients.
5.8 Therapy
Neuroblastoma patients younger than 6 months with small adrenal masses have an excellent event-free survival and overall survival rate (>95 %) without surgical intervention [196]. Low-risk patients with International Neuroblastoma Staging System (INSS) 1 and 2 and International Neuroblastoma Risk Group Staging System (INRGSS) L1 whose tumors have no unfavorable features, the outcome is excellent with surgery alone, and immediate use of chemotherapy may be restricted to a minority of patients with low-risk patients [197, 198]. Intermediate-risk neuroblastoma patients with tumors that are unresectable at diagnosis (INRGSS L2 or INSS 3 tumors), who received dose-intensive chemotherapy, have an excellent overall survival rate (>80 %) [199, 200]. Further studies demonstrated that duration of chemotherapy and doses of chemotherapeutic agents could be reduced to prevent side effects [200, 201]. High-risk neuroblastoma patients mainly include stage 4, > 18 months of age, and stage 3 with MYCN amplification or stage 3 > 18 months with unfavorable histology. Additionally, for children with metastatic disease or unresectable tumors with MYCN amplification, the outcome remains poor despite improvements in survival after primary tumor resection, multiagent chemotherapy, irradiation, anti-GD2 immunotherapy, and/or 13-cis-retinoic acid treatment [6, 202–204]. Despite these advances, the overall survival rate of high-risk neuroblastoma patients is still <50 %, and 5-year overall survival of patients with relapse disease is only 8 % [202, 205]. Thus, research into novel, rationally designed therapies that target high-risk neuroblastoma patients is urgently required. Figure 5.2 displays an overview of the therapeutic targets in neuroblastoma based on recent genetic and epigenetic studies.
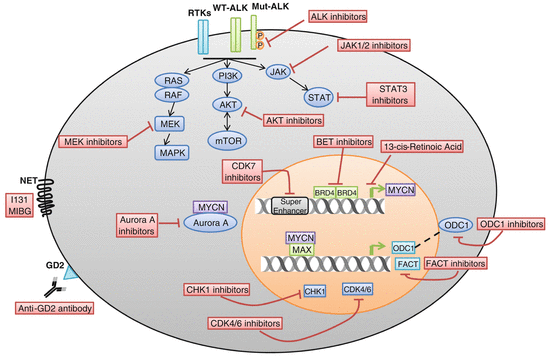
Fig. 5.2
Therapeutic targets in neuroblastoma. (i) Receptor tyrosine kinases (RTKs), wild-type ALK (WT–ALK) or mutant ALK (Mut–ALK), as well as downstream RAS–MAPK, PI3K/AKT/mTOR, and JAK/STAT pathways are aberrantly activated in neuroblastoma; thus, they are therapeutic targets in neuroblastoma. (ii) MYCN is an oncogenic driver in neuroblastoma, and compounds that can inhibit MYCN transcription, destabilize N-Myc protein, or inhibit its downstream mediators are potential drugs for neuroblastoma therapy. (iii) Cyclin-dependent kinases CDK4/6 and checkpoint proteins CHK1 in neuroblastoma are aberrantly activated; thus, they are therapeutic targets for neuroblastoma. (iv) GD2 is overexpressed in almost all of the neuroblastomas yet shows restricted normal tissue expression making it a good target for immunotherapy of neuroblastoma. (v) Norepinephrine transporter (NET) is expressed in 90 % of neuroblastoma tumors, and NET is able to uptake radiopharmaceutical 131I-metaiodobenzylguanidine (131I-MIBG); thus, 131I-MIBG is used for irradiation targeted therapy of neuroblastoma
5.8.1 Anti-GD2 Immunotherapy
Surface glycolipid molecule disialoganglioside (GD2) is overexpressed in pediatric and adult solid tumor such as neuroblastoma, glioma, retinoblastoma, sarcomas, and melanoma [206]. However, GD2 expression level is low in normal tissues and restricted to neurons, melanocytes, some nerve fibers, and basal layer of the skin [206–208], which makes GD2 a suitable target for cancer immunotherapy. GD2 is associated with cancer cell proliferation, migration, and invasion, presumably via activation of the receptor tyrosine kinase (RTK) pathway or other tyrosine kinases [209–213]. The first developed anti-GD2 monoclonal antibodies (mAb) were murine anti-GD2 mAbs including 3F8, 14G2a, and ME36.1 [214–216]. Phase II studies showed antitumor effects of the anti-GD2 mAbs in high-risk neuroblastoma patients [217]. To reduce the human anti-mouse antibody levels in the serum, chimeric (murine x human) and humanized anti-GD2 mAbs were developed. Chimeric 14.18 (ch14.18) is one such antibody. In a phase III study, a total 226 eligible patients were randomly assigned to a treatment group. One hundred thirteen patients received standard therapy (isotretinoin), and a similar cohort received immunotherapy (isotretinoin combined with GD2 antibody ch14.18, GM-CSF, and IL-2). The results showed that the immunotherapy group had significantly higher event-free survival (66 ± 5 % vs. 46 ± 5 % at 2 years, P = 0.01) and overall survival compared to control group (86 ± 4 % vs. 75 ± 5 % at 2 years, P = 0.02 without adjustment for interim analyses). This indicates that the cohort receiving immunotherapy had improved outcomes compared with standard therapy in high-risk neuroblastoma patients [218]. However, the role of anti-GD2 immunotherapy in the relapse setting and patients with bulky disease needs to be further investigated. In addition to anti-GD2 IgG mAbs, other studies that target GD2 are ongoing, which include the generation of chimeric antigen receptor (CAR)-modified T cells, radiolabeled antibodies, drug conjugates, and nanoparticles [206].
5.8.2 131I-metaiodobenzylguanidine Therapy
Norepinephrine transporter (NET) is expressed in 90 % of neuroblastoma tumors, and NET is able to uptake radiopharmaceutical 131I-metaiodobenzylguanidine (131I-MIBG), which is used to image disease burden in neuroblastoma patients and is currently being evaluated for its therapeutic potential [219, 220]. Clinical trials in relapsed neuroblastoma showed positive responses albeit with severe side effects due to myelosuppression, which could be abrogated by hematopoietic stem cell transplant [221–223]. While the majority of neuroblastomas are avid for 131I-MIBG on imaging, only 30–40 % of patients have partial or complete responses [220, 221], suggesting combination treatment may be required to improve the efficacy of 131I-MIBG therapies. In fact, a clinical trial using 131I-MIBG in combination with chemotherapy showed promising results: in a phase II study, combination of 131I-MIBG with the chemo-drug topotecan in newly diagnosed high-risk neuroblastoma patients leads to an overall objective response rate of 57 %, and a response of 94 % in the primary tumor indicates that it is an effective treatment option for these patients [224].
5.8.3 Targeting ALK
In neuroblastoma, ALK is the most commonly mutated gene (8 %), with an additional 3 % harboring ALK amplification [87–91]. Crizotinib is an ATP-competitive dual-specific inhibitor of ALK/c–Met that has been approved for treatment of ALK-rearranged non-small-cell lung cancer [225]. The treatment of neuroblastoma cells with crizotinib inhibits proliferation of neuroblastoma cell lines expressing either R1275Q-mutated ALK or amplified wild-type ALK, but is not effective for cell lines harboring the F1174L-mutated ALK. The crizotinib resistance of ALK 1174 L tumors is due to a failure of crizotinib to bind to the ALK ATP-binding pocket [91, 226, 227]. In a phase I study of crizotinib in 34 neuroblastoma patients (11 with known ALK status), only one patient had a complete response, while two had stable disease [228]. Recently, several new and more potent ALK inhibitors such as ceritinib (LDK378), alectinib (CH5424802), AP26113, RXDX-101, and PF-06463922 have emerged and are in preclinical studies [229]. The next-generation ALK/ROS1 inhibitor PF-06463922 showed excellent activity in ALK F1174L neuroblastoma models that were resistant to crizotinib [230]. In addition to small molecules, an antibody targeting ALK has been found to inhibit neuroblastoma cells growth with either wild-type or mutated ALK, suggesting that ALK-targeted immunotherapy is a promising therapeutic strategy for neuroblastoma [231].
5.8.4 Targeting MYCN
MYCN oncogene amplification is associated with aggressive neuroblastoma and occurs in approximately 20 % of primary neuroblastoma tumors and approximately 40 % of high-risk neuroblastomas. Although it is an oncogenic driver in neuroblastoma, it appears that the elevated levels of MYCN may give tumors with this genetic alteration an “Achilles heel” making them more sensitive to a variety of therapeutic approaches. Thus targeting MYCN has great potential for high-risk neuroblastoma therapy. One of the early attempts to target MYCN evolved from the finding that retinoids inhibited MYCN transcription resulting in tumor cell growth arrest and differentiation [232]. Later retinoids were integrated into the consolidation phase of high-risk patient therapy and resulting in an increase in overall survival [233]. Blockade of MYC-MAX dimerization is a proposed mechanism to inhibit MYC transcriptional activity [234]. Recent studies showed that a characterized c-MYC/MAX inhibitor 10058-F4 also targets MYCN/Max interaction, which leads to suppression of MYCN amplified neuroblastoma cell proliferation and an increase in MYCN transgenic mice survival [235]. Recent preclinical studies suggest that bromodomain and extraterminal domain (BET) inhibitors inhibit MYCN transcription and induce cell death [236, 237]. Like some other critical oncogenes, it has been shown that the transcription of MYCN is driven by a super enhancer [238, 239] and a covalent inhibitor of cyclin-dependent kinase 7 (CDK7) disrupts the transcription of MYCN as well as other super-enhancer-associated genes, leading to the tumor regression in a mouse model of high-risk neuroblastoma [238]. Activation of the phosphatidylinositol 3’-kinase (PI3K) signal transduction pathway and Aurora A kinase act at various levels to block the N-Myc protein degradation pathway and contribute to high N-Myc protein levels in tumor cells [240–242]. PI3K and Aurora A kinase inhibitors destabilize N-Myc protein and inhibit neuroblastoma tumor growth [240–242]. Identifying the requisite biopathways downstream of MYCN can also provide therapeutic opportunities. ODC1, the rate-limiting enzyme in polyamine biosynthesis, and histone chaperone FACT (facilitates chromatin transcription) are found to be crucial mediators of the MYCN signal. ODC1 or FACT inhibitor treatment suppresses tumor growth in a mouse model of high-risk neuroblastoma (MYCN transgenic mouse model) [36, 37]. These findings indicate small molecules that target MYCN transcription, destabilize N-Myc protein stability, or inhibit MYCN downstream mediators may have great potential for the treatment of high-risk neuroblastoma patients.
5.8.5 Targeting RAS–MAPK
WGS identified clonally enriched somatic mutations that are predicted to activate the RAS–MAPK pathway in 78 % of relapsed neuroblastoma tumors [127], making the RAS–MAPK a good target for treating relapsed high-risk neuroblastoma therapy. Neuroblastoma cell lines with mutations in RAS–MAPK pathway but not wild-type RAS–MAPK are sensitive to MEK inhibitors in preclinical xenograft mouse models suggesting a role for these agents in the relapsed disease [127]. Loss of the tumor suppressor NF1 activates RAS–MEK signaling in neuroblastoma cells and makes the cells resistant to retinoic acid-induced cell differentiation, which can be overcome by addition of MEK inhibitors [243]. These data indicate that the combination of MEK inhibitor and retinoic acid treatment may be beneficial to patients with high-risk neuroblastoma.
5.8.6 Targeting Receptor Tyrosine Kinases and PI3K–AKT
Neuroblastoma patients with high brain-derived neurotrophic factor (BDNF) and its receptor tyrosine kinase TrkB have an unfavorable prognosis, and activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis through PI3K/AKT pathway [244–247]. Immunohistochemical staining of neuroblastoma samples showed that phosphorylation of Akt occurred in the majority of the samples and is associated with reduced event-free and overall survival [248]. The aberrant activation of PI3K/AKT/mTOR pathway was also found in high-risk neuroblastoma patients by others [249]. The PI3K/AKT/mTOR pathway drives the malignant process in neuroblastoma partially due to MYCN stabilization [242, 250, 251]. Many preclinical studies have been performed using compounds against PI3K, AKT, and mTOR. PI3K inhibitor such as NVP-BKM120 alone or in combination with chemotherapeutics has been show to inhibit neuroblastoma cell growth [252, 253]. In preclinical studies, AKT inhibitors such as perifosine alone or in combination with chemotherapeutics suppress neuroblastoma growth [254–256]. MTOR inhibitors such as rapamycin or temsirolimus have been shown to inhibit neuroblastoma growth in vitro and in vivo in preclinical studies [257–259], but clinical studies showed little activity in the Phase II setting [260]. Recent preclinical studies have shown that MYCN amplified tumors may be sensitive to the combination of the allosteric AKT inhibitor MK2206 and rapamycin by preventing the feedback activation of AKT or mTOR when either agent is used alone [256]. Taken together, inhibition of the PI3K/AKT/mTOR pathway alone or in combination with standard therapy is a promising approach for high-risk neuroblastoma.
5.8.7 Targeting JAK–STAT3
The JAK/STAT pathway is implicated in the pathogenesis of many human cancers. AZD1480 is a pharmacological JAK1/2 inhibitor. AZD1480 treatment blocks endogenous and IL-6-induced STAT3 activity and suppresses neuroblastoma cell growth in vitro and in neuroblastoma xenografts [261]. Moreover, in neuroblastoma, a highly tumorigenic subpopulation termed cancer stem cell (CSC) has been shown to express CSF3R, a receptor for granulocyte-colony-stimulating factor (G–CSF), and the receptor ligand G–CSF activates STAT3 in neuroblastoma CSC subpopulations [262]. Blockade of this G–CSF–STAT3 signaling loop with either anti-G–CSF antibody or STAT3 inhibitor depleted the CSC subpopulation within tumors, resulting in tumor growth inhibition, decreased metastasis, and increased chemosensitivity [263]. Thus, inhibition of JAK/STAT signal transduction could be a promising therapeutic target for primary neuroblastoma as well as relapsed neuroblastoma enriched with CSCs.
Related posts:
 Molecular Genetics of Gastroenteropancreatic Neuroendocrine Tumours
Molecular Genetics of Gastroenteropancreatic Neuroendocrine Tumours
 Molecular Genetics of MEN1-Related Neuroendocrine Tumors
Molecular Genetics of MEN1-Related Neuroendocrine Tumors
 Radionuclide Imaging of Pancreatic Neuroendocrine Tumours
Radionuclide Imaging of Pancreatic Neuroendocrine Tumours
 Nanoparticles for Radionuclide Imaging and Therapy: Principles
Nanoparticles for Radionuclide Imaging and Therapy: Principles
 Radionuclide Imaging of Medullary Thyroid Carcinoma
Radionuclide Imaging of Medullary Thyroid Carcinoma
 Radionuclide Imaging of Gastrointestinal Neuroendocrine Tumors
Radionuclide Imaging of Gastrointestinal Neuroendocrine Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


