Fig. 7.1
HCC evolution and its related molecular and immunological markers. CLD chronic liver disease, RN regenerative nodule, DN dysplastic nodule, N/N nodule-in-nodule [28]
7.4.1 Heat Shock Protein 70 (HSP70)
HSP70 is a family of 70-kDa chaperone stress proteins that assists in a wide range of protein folding processes, including the folding and assembly of newly synthesized proteins, refolding of misfolded, denatured, or aggregated proteins, and the membranous translocation of proteins [33]. HSP70 is a potent antiapoptotic cellular survival factor, whose overexpression promotes cell cycle progression, and plays a role in tumorigenesis, with high expression levels associated with adverse outcomes in endometrial, colorectal, breast, prostate cancers and some leukemias [34–39]. Chuma et al. [40] analyzed the expression of ~12,600 genes in seven nodular HCCs and seven corresponding noncancerous hepatic tissue. HSP70 was overexpressed in early HCC compared to most high-grade dysplastic nodules, and was further increased in high-grade HCC. Interestingly, non-malignant/benign nodules, focal nodular hyperplasia, and hepatocellular adenomas showed low HSP70 expression [40, 41]. HSP70 expression is not entirely specific for HCC, as ~5 % of high-grade dysplastic nodules overexpress this protein [41].
7.4.2 Bmi-1
Bmi-1 is identified as a stem cell gene based on the observation that Bmi-1 deficiency compromises adult stem cell function [42]. Bmi-1 encodes a 37 kDa member of the Polycomb gene group which imparts stem cell features to malignant cells, including the induction of telomerase activity and the negative regulation of the p16 and p19 tumor suppressor genes [43, 44]. Bmi-1 overexpression correlates with therapy failure in many different human malignancies [45–47]. Interestingly, down regulation of BMi-1 results in apoptosis and senescence, and in increased cellular sensitivity to cytotoxic agents [48]. Bmi-1 expression is increased in early and well-differentiated HCC usually appearing as small dots within the nucleus. In one study 65 % of well differentiated, 45 % of moderately differentiated, and 50 % of poorly differentiated HCCs expressed Bmi-1 [49].
7.4.3 Glutamine Synthetase (GS)
GS is found predominantly in the brain, kidneys, and liver, where plays a central role in nitrogen metabolism by catalyzing the condensation of ammonia and glutamate to glutamine [50]. Interestingly, glutamine is known to act as an energy source for tumor cells [51]. In normal liver GS expression is found in the hepatocytes surrounding the terminal hepatic venules, while in HCC there is diffuse and extensive accumulation of GS in the malignant hepatocytes [52]. One study demonstrated increased GS expression in 14 % of high-grade dysplastic hepatic nodules as compared to 59 % of well-differentiated HCCs, and 86 % of moderately to poorly differentiated HCCs [41]. GC overexpression is associated with β-catenin mutations or activation of the β-catenin pathway [53, 54]. Interestingly, increasing GS expression appears to promote HCC progression and metastasis [55, 56]. GS is not entirely specific as increased expression is seen in 14 % of high-grade dysplastic hepatic nodules and in the β-catenin-activated hepatocellular adenoma subtype [41, 57].
7.4.4 α-Fetoprotein (AFP)
AFP is a well established serum marker for HCC, with ~50 % of individuals with HCC showing elevated AFP and plasma levels above 400–500 ng/ml AFP being virtually diagnostic of HCC [58]. Unfortunately APF levels are seldom elevated in early HCC and it has low sensitivity as a tissue marker, being expressed in only about 17–68 % of paraffin embedded HCC sections [58, 59]. In addition, AFP levels are usually not elevated in the fibrolamellar subtype of HCC.
7.4.5 Cyclase-Associated Protein 2 (CAP2)
7.4.6 Glypican-3 (GPC3)
GPC3 is a cell-surface heperan sulfate proteoglycan normally expressed in fetal liver and placenta, but not in normal adult liver. Recently, GPC3 has been established as a serum and tissue maker for HCC. One recent study demonstrated GPC3 immunoreactivity in 69 % of well differentiated and 81 % of moderately to poorly differentiated HCCs. Only 9 % of high-grade dysplastic nodules and no low-grade nodules were positive [41]. GPC3 immunoreactivity shows 77 % sensitivity and 96 % specificity for small HHC, demonstrating that positivity for this immunostain can support the diagnosis of HCC. GPC3 typically shows a cytoplasmic staining pattern, although canicular or membranous patterns are also seen [41, 63, 64]. GPC3 immunopositivity needs to be interpreted in the correct clinical and histological context, as benign liver lesions with active inflammation and regeneration, and melanocytic lesions can also be GPC3 immunopositive [65, 66].
7.4.7 Immunostain Combinations for HCC
Like for immunohistochemical procedures, various immunostain combinations give better result in the diagnosis of HCC then a single stain. The combination of GPC3, HSP70, and GS allowed 72 % sensitivity and 100 % specificity in well differentiated HCC detection when two immunostains were positive. GPC3 and HSP70 alone gave a 59 % sensitivity and 100 % specificity for well-differentiated HCC. These results are significantly better than those achieved with any single immunostain [41].
7.5 Molecular-Genetic Testing for HCC
Currently, there are relatively few molecular-genetic tests routinely performed for HCC. Presently the Association for Molecular Pathology Solid Tumors Test Directory lists p53 mutational analysis for HCC, offered by two labs and no other tests [67]. However, there are several important molecular tests related to HCC risk and several areas of molecular pathology research that are likely to lead to new molecular diagnostic tests for HCC.
7.6 Hereditary Hemochromatosis
HH is one of the most common genetic diseases, affecting about one in 200–300, with Northern Europeans showing the highest known incidence. HH increased an affected individuals HCC risk, with different HH types often conferring a different risk. Presently there are five different known molecular causes of HH. Since untreated HH can result in cirrhosis and HCC, and can be inherited, molecular testing for HH followed by correct clinical care is necessary [10]. Presently, molecular diagnostic tests are available and routinely preformed in many labs, with HFE gene mutation being the most common.
7.6.1 Indications for HH Testing
The initial diagnosis of HH can be difficult due to the protean manifestations of HH. The symptoms of HH can include heart failure/cardiac arrhythmias, cirrhosis, diabetes, arthritis, joint pain, skin bronzing, fatigue, hypogonadism, hypothyroidism, and HCC [9–11]. It is important to identify individuals with HH as early as possible. While some manifestations of HH can be reversed by therapeutic phlebotomy, such as the liver and skin manifestations, other complications, such as hypogonadism, arthropathy, and diabetes, are irreversible once they have occurred [68]. Should an individual have a family history of HH, molecular testing is warranted to avoid missing HH in its earliest and most treatable form [10, 68]. Evaluation of iron overload should first consider non-genetic causes, many of which are more common than HH.
Iron overload can be measured by several tests, including total iron binding capacity, transferrin saturation, liver biopsy, and serum ferritin (SF). Initial testing is often performed by serum ferritin analysis, which is highly sensitive for iron overload, showing hyperferritinemia. Care must be taken in interpreting the test result, as SF is an acute phase reactant and hyperferritinemia also occurs in inflammatory processes, diabetes, chronic alcohol use, and liver damage [10]. Testing should be performed to exclude other disease processes. Santos et al. [69] proposed the following four-step strategy:
1.
Rule out other causes of hyperferritinemia. Taking a personal and family history, performing a clinical exam, and performing tests such as iron parameters, C-reactive protein, hemoglobin, and alanine aminotransferase and aspartate aminotransferase will usually give a diagnosis of acquired hyperferritinemia and avoid unnecessary molecular testing.
2.
Confirm iron overload. Since SF levels can vary throughout the day, repeating SF can be useful. Along with other iron studies and diagnosis of iron overload can be rendered. Possible causes of iron overload include excessive iron supplementation (oral, parenteral, or transfusions), chronic liver disease (alcoholic, viral or metabolic), end stage liver disease, chronic anemia (thalassemia major, myelodysplastic syndrome, sideroblastic anemia, chronic hemolysis), and porphyria cutanea tarda.
3.
Assess hepatic iron stores. This can be done via liver biopsy or magnetic resonance imaging. Hepatic iron store analysis may be necessary where other iron studies fail to give definitive results.
4.
Perform molecular genetic testing for HH. While mutations of the HH gene are most common, other genetic causes of HH must also be considered. An algorithm for molecular genetic testing protocols is given in Fig. 7.2.
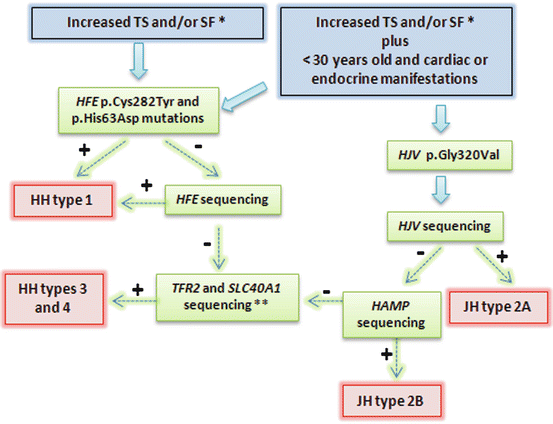
Fig. 7.2
Representation of diagnostic strategy for patients suspected HH. Recommendations report TS > 45 %, SF > 200 μg/L in females and >300 μg/L in males; or in advanced stages: TS > 50 % in females and TS > 60 % in males, in the absence of secondary causes. Some patients with primary iron overload may not present mutation during this genetic approach. Very rare mutations in other genes can be involved. TS transferrin saturation, SF serum ferritin, JH juveline hemochromatosis. + means positive result, and − means negative result; lane 7, heterozygous for C282Y, wild-type for H63D; lane 8, wild-type for both C282Y and H63D [10]
7.6.2 HFE Testing
The gene encoding HFE is located at 6p21.3 and encodes a seven exon, six protein domains, 47.5 kDa protein similar in structure to the major histocompatibility class I-like proteins. The exoms encode an initial signal sequence, three extracellular domains, a transmembrane region and a short cytoplasmic tail, forming a 4.2 kb long transcript. The seventh exon does not appear to be expressed [70]. HFE is mainly expressed in the liver, duodenum, small intestine, spleen, and heart. Several alternatively spliced variants are expressed at low levels in many human tissues [70–76]. Most cases of HH are caused by two single nucleotide polymorphisms (SNP), Cys282Tyr and His63Asp, making the inheritance autosomal recessive. Some 20 other less common HFE mutations are linked to HH, which contribute to the phenotypic heterogeneity of the disease. Most commonly, individuals with HH are homozygous for Cys282Tyr, or heterozygous for Cys282Tyr/His63Asp [10]. In Northern European populations, roughly 90 % of HFE mutations are Cys282Tyr, with Cys282Tyr/Cys282Tyr being the most common HH genotype. Homozygousity for His63Asp does not cause HH, but this SNP contributes to increased hepatic iron levels when the Cys282Tyr SNP is also present [76]. HFE mutation causes excess iron accumulation via reducing the amount of hepcidin levels, resulting in increase ferroportin activity and increased enteric iron import [77].
HFE testing for HH is performed employing a variety of different molecular tests. Usually the testing modalities include a combination of PCR amplification, restriction enzyme digest, allele- specific PCR, heteroduplex analysis, sequencing, and single-strand conformation polymorphism testing. Here a few of these techniques are reviewed.
7.6.2.1 Mutliplex PCR with Restriction Endonuclease Digestion
Stott et al. [78] employed a combination of mutliplex PCR with primer mismatches, followed by restriction endonuclease digestion to identify the Cys282Tyr and His63Asp mutations in peripheral blood samples. Five milliliter of blood was taken via venipuncture and the DNA was extracted. The DNA was then PCR amplified with the primers depicted in Fig. 7.3a. Note that the Cys282Tyr and His63Asp each had a different set of mismatched primers that introduced a BbrPI restriction site into the wild type, but not in either mutant amplicon. The amplification was carried out under standard conditions with a 2 min denaturation at 96 °C, followed by an initial annealing step of 30 s at 55 °C, with 30 cycles each of 30 s at 72 °C, 30 s at 94 °C, and 15 s at 55 °C. A final extension period of 10 min at 72 °C completed the PCR.
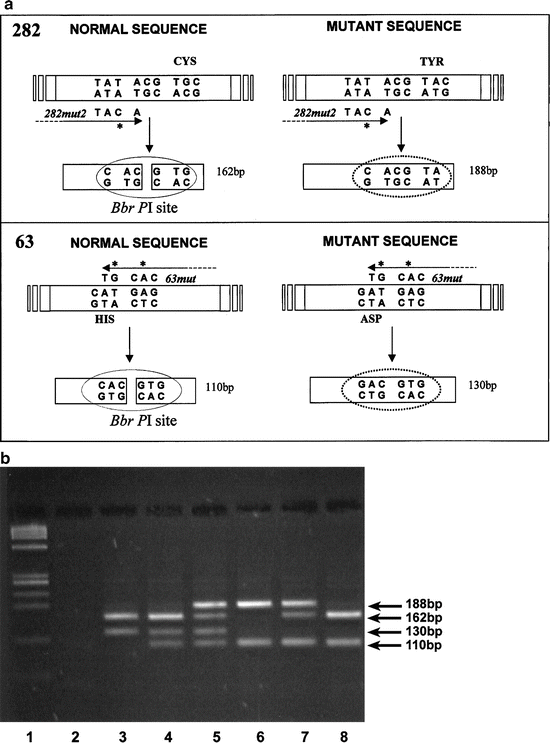
Fig. 7.3
(a) Schematic outline of PCR and restriction digestion used to determine the presence of HFE gene mutations and detection of both hemochromatosis mutations, C282Y and H63D, by PCR and restriction digestion (b). The site of mutation and primer mismatches are shown for the 282 locus (top) and for the 63 locus (bottom). (b) Restriction fragments were separated on 3 % Nusieve:Agarose. Results representative of all six possible genotypes are shown. Lane 1, PHI-X174-HaeIII molecular mass marker; lane 2, negative control; lane 3, wild-type for C282Y, homozygous for H63D; lane 4, wild-type for C282Y, heterozygous for H63D; lane 5, heterozygous for C282Y, heterozygous for H63D (compound heterozygote); lane 6, homozygous for C282Y, wild-type for H63D [79]
The entire PCR reactions were then subjected to BbrPI restriction digestion for 4 h using five Units of enzyme. The restriction fragments were separated by gel electrophoresis using 3 % Nusieve:Agarose (3:1) gels for 50–60 min at 100 V. Products were visualized by staining in 1 mg/L ethidium bromide followed by transillumination at 302 nm. Each possible combination of the two SNPs and wild type sequence are represented in Fig. 7.3b. Each combination gave a specific and reproducible pattern that allowed identification of the two common HFE SNPs.
7.6.2.2 Allele-Specific PCR (ARMS)
ARMS takes advantage of the 10−3 to 10−6 lower amplification efficiency of the Taq DNA polymerase initiating from mismatched template-primer 3′-termini [79]. Through the use of primers specific for either a wild type or SNP sequence, it is possible to design primers that will preferentially amplify the sequence they share 100 % homology with [80]. In some systems ARMS technology can detect one mutant allele in a background of 104 wild-type alleles [81]. Sánchez et al. [81] used ARMS to analyze the blood of 5,370 Spanish blood donors for the Cys282Tyr and His63Asp HFE mutations. DNA was isolated via cell lysis, high-speed centrifugation, multiple washing steps, and protein removal by proteinase K digestion [82, 83]. The DNA was subjected to PCR amplification with the following probes:
Cys282Tyr | His63Asp |
|---|---|
C282Y-R1: CCACTGATGACTCCAATGACTA | 63Fw: AGCTGT TCGTGTTCTATGATC |
C282Y-Rm: CCTGGGTGCTCCACCTGGT | HFE187Gas: CTCCACACGGCGACTCTCATC |
C282Y-Fw: GGGAAGAGCAGAGATATACGTG | HFEEx2U: GCTCCCCTCCTACTACACAT |
C282Y-F: TGGCAAGGGTAAACAGATCC | HFEEx2L: GGTCCCTATTTCCACCATCC |
The allele-specific PCR was preformed under standard conditions for 35 cycles and the resulting amplicons were analyzed. Of the 5,370 blood samples analyzed, 8 C282Y homozygotes (0.15 %) and 74 compound C282Y/H63D heterozygotes (1.38 %) were identified, giving a C282Y allele frequency in the Spanish population of 3.16+/−0.46 %, or 1 homozygote in every 1,004 individuals. The H63D homozygote frequency was 20.8 %, or 1 in every 23 individuals. The allelic frequencies were not significantly different between men and women. To confirm that the above assay worked, the researchers employed the assay by Stott et al. [78] and found a 100 % results concordance. Four of the 8 C282Y homozygotes, all men, had high serum ferritin and transferring iron saturation, while only 1 of the 74 C282Y/H63D heterozygotes had the same serum results, demonstrating that the compound heterozygotes were less likely to develop HH.
7.6.2.3 DNA Sequencing
AHH is characterized by a number of common SNPs and in most cases analysis of these SNPs by techniques, such as ARMS, will locate specific HFE gene mutations. However, since HH can be caused by loss of HFE expression, resulting in increased ferroportin activity [10], other less common mutations that result in the loss of HFE protein expression can also cause HH. Sequencing is the ideal technique to locate these HFE gene variants, as it gives the specific nucleotide type at each point interrogated by the molecular assay. Cukjati et al. [83] analyzed a 47-year-old man, with no family history of HH, who had repeated high serum iron levels without any history of chronic anemia, infectious liver disease, alcohol abuse, or oral or parenteral iron administration. The patient’s blood and the blood from 22 Slovenian patients with HH was collected and the DNA was extracted using a QIamp DNA Blood Mini Isolation Kit. RT-PCR analysis for the common HFE mutations (C282Y, H63D, and S65C) was performed on these blood samples. Of the 22 Slovenian patients with HH, four did not carry any of the common HFE gene SNPs tested for above. Additionally, the patient and his son were negative for all three common HFE gene SNPs.
To further analyze this set of patients, exons 1 → 6 with PCR amplified and subjected to Sanger sequencing using an ABI PRISM Big Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems) and analyzed on an ABI PRISM 310 Gene Analyzer. In three patients with HH, no mutations were identified by this analysis method, while the 47-year-old had a frameshift homozygous mutation of the A nucleotide in exon 3, at position 471 (c.471del, p.A158fs), which resulted in a prematurely truncated protein. The patient was also homozygous at both HLA-A and HLA-B, suggesting that loss of heterozygosity might have occurred during mitotic recombination. While rare, unusual HFE gene mutations require testing by sequencing or other molecular techniques capable of interrogating every nucleotide within the DNA sequence being analyzed. Although not performed often, HFE gene mutations have also been analyzed by DNA pyrosequencing with good results [84].
7.6.2.4 Genotyping HFE Gene Mutations by Melting Point Analysis
DNA melting point analysis typically involves PCR amplification followed by allele-specific fluorescent probe melting profile analysis. This technique is commonly employed to locate changes in nucleic acid sequences, although by itself it does identify specific nucleotide changes. It has the advantage of allowing melting analysis to be performed on the closed PCR reaction without the need of opening the reaction tube once amplicon amplification is complete. This significantly lowers the chances of sample contamination.
Bach et al. [85] used this technique to analyze genomic DNA from over 3,500 donor samples to look for the presence of the C282Y (845G > A), H63D (187C > G), and S65C (193A > T) HFE gene mutations. The mutations were amplified with the following primers:
C282Y | H63D and S65C |
|---|---|
F5′-TGGCAAGGGTAAACAGATCC-3′ | F5′-GCTCTGTCTCCAGGTTCACACTC-3′ |
R5′-CTCAGGCACTCCTCTCAACC-3′ | R5′-CCCTCTCCACATACCCTTGC-3′ |
The amplification was performed as follows: denaturation 1 cycle 95 °C 10 min, followed by 45 cycles of 95 °C 10 s, 55 °C 10 s, and 72 °C 10 s, with a temperature transition rate of 20 °C/s. The melting curve was performed at 95 °C for 5 s and 45 °C for 15 s with a temperature transition rate of 20 °C/s ramping to 85 °C at 0.1 °C/s. The C282Y/845A > T transition was measured with a 3′-fluorescein-labeled sensor (5′-GAG ATA TAC GTG CCA GGT GGA GCA C-3′) and a 5′-LC-Red640-labeled anchor probe (5′-AGG CCT GGA TCA GCC CCT CAT TGT-3′). The H63D/187C > G was monitored with a 5′-LC-Red705-labeled anchor (5′-CTG GTA TCC ACG TAG CCC AAA GCT TCA A-3′) and a 3′-fluorescein-labeled sensor (5′-GG CGA CTC TCA TGA TCA TAG AAC ACG AAC A-3′) probe. This second probe- anchor combination was also used to monitor the S65C/193A > T mutation as the SNP was located only six nucleotides away from the H63D/187C > G mutation. Abnormal melting curves were analyzed by Sanger sequencing on an ABI PRISM 310 Gene Analyzer. By this method two new HFE gene substitutions were identified and one melting curve typical of the C282Y/845A > T transition was actually G > A transition located two nucleotides away from the C282Y/845A > T transition, giving the same melting curve. The authors concluded that this analysis method works well, but in some cases results should be further analyzed by DNA sequencing or restriction endonuclease digest.
7.6.3 Juvenile Hemochromatosis (JH)
JH or Type 2 HH is a rare autosomal recessive disorder that leads to significant iron overload before the age of 30 and is characterized by cardiomyopathy, liver damage, endocrine dysfunction, hypogonadotrophic hypogonadism. JH typically has a rapidly progressive clinical course, making the rapid initiation of therapy vital for patient care and also allowing the clinical differentiation from HFE mutation-induced HH (see Fig. 7.2). Molecular analyses have demonstrated that two genes are mutated in JH; HJV and HAMP, leading to JH types 2A and 2B, respectively. The HJV gene consists of four exons, encoding the hemojuvelin protein Individuals with HJV gene mutations and HJV knockout mice have low hepcidin, suggesting that hemojuvelin is involved in regulating hepcidin expression. The HAMP gene encodes three exons and encodes hepcidin, a peptide hormone produced in hepatocytes that plays a role in iron absorption and ferroportin degradation in intestinal enterocytes [10].
7.6.3.1 HJV Gene Analysis
The HJV gene has a large number of mutations that lead to JH 2A, with the most common one being a Gly320Val change [86–89]. Due to the rarity of HVJ gene mutations and the relatively large number of mutations that can cause type 2A JH, sequencing has often been chosen to analyze JHV gene mutations. Lee et al. [89] employed these primers to amplify the HJC gene exons in unrelated kinships in the Southeastern US in which HJ had been identified:
HJV | Primer sequence | Fragment size in BP |
|---|---|---|
HJ Ex 1 F | GTACTCTGGCCAGCCATATACT | 286 |
HJ Ex 1 R | CGAGAGACATCCAAGTAGGTGT | |
HJ Ex 2 F | ATCTCCCCAAATTCCAGTCTG | 359 |
HJ Ex 2 R | ACATAGCAGCCTACCCTCTAG | |
HJ Ex 3 F | GCAAACTACACTCCGATAGAG | 669 |
HJ Ex 3 R | GAATCTCATGAGGTGGATCGG | |
HJ Ex 4A F | TAGTCCTGCATCTCTACTTGG | 394 |
HJEx4AR | TGCAGGTCCTGTTCAGCTG | |
HJ Ex 4B F | ATGGAGGTGACCGACCTGG | 374 |
HJEx4BR | AGCTGCCACGGTAAAGTTGG | |
HJ Ex 4C F | GCTCTCCTTCTCCATCAAGG | 430 |
HJEx4CR | AAACTAGTAATGGGACTGATGG | |
HJ Ex 4D F | TGTGGGCTCTTTGTTCTGTG | 407 |
HJEx4DR | GTCTTCTGCTTTCAGCTCTTG | |
HJ Ex 4E F | ATAAGTTTAGAGGTCATGAAGG | 404 |
HJEx4ER | GCCCTCTTTCAGTGGAGTG |
The amplicons were purified using a Qiaquick PCR purification kit and the genotypes were determined by direct sequencing with an ABI 3100 DNA sequencer. By this method first group had two novel HVJ gene mutations, while to second group had two previously identified mutations.
7.6.4 Transferrin 2 Gene Mutation (TFR2) and Hemochromatosis
TFR2 gene mutations cause Type 3 HH, an HH type with similar clinical manifestations the HFE mutation-induced HH [10, 92]. The TFR2 gene encodes an 18 exon, 801 amino acid protein that shares 45 % identity with TFRC. The protein mediates cellular iron import and has two predominant transcripts (alpha and beta), with the alpha form expressed mainly in the liver, spleen, lung, muscle, prostate, and peripheral blood mononuclear cells, and the beta form expressed in all tissue examined [92, 93]. Once identified, Type 3 HH is treated with periodic phlebotomies, like most other HH types [10].
TFR2 genes mutations leading to HH are relatively rare [10]. Roetto et al. [94] examined four different families with HH unrelated to HFE gene mutations. DNA was prepared from peripheral blood by standard phenol-chloroform extraction. The DNA was PCR amplified in a Thermal Cycler using 10 pmol of primers specific to all 18 TFR2 exons. A total of 32 PCR cycles (denaturation, 94 °C for 30 s; annealing, 56 °C for 45 s; extension, 72 °C for 45 s) were performed. The amplicons were then transcribed into RNA with 10 units T7 RNA polymerase in an appropriate buffer with 35S-UTP and subjected to gel etectrophoresis, drying, and autoradiography. Bands showing altered electrophoretic mobility were directly sequenced. Direct sequencing was performed on the purified PCR amplicons using a Thermo Squenase Cy5.5 dye terminator cycle sequencing kit. After removal of the unincorporated dye, the sequence was identified via electrophoresis on an automatic sequencer (model 373A; Applied Biosystem). The MaeI enzyme was used to detect the previously described Y250X mutant within the amplicon for exon 6.
Of the patients with altered TFR2 gene exon electrophoretic mobility two patients had a previously described Y250X mutation in exon 6. In one large inbred family a frameshift mutation (84–88 insC; E60X) in exon 2 was identified which formed a premature stop codon. A single patient with nonfamilial HH carried a T → A transversion (T515A). All patients with a non-protein expressing phenotype showed severe clinical complications.
In another study, Biasiotto et al. [92] used denaturing high-performance liquid chromatography to analyze TFR2 gene mutations using blood samples from 657 individuals from Northern Italy who either had a diagnosis of HH or serum ferritin concentrations greater than 400 μg/L or a transferrin saturation above 50 %. The HFE gene C282Y and H63D mutations were identified by endonuclease digestion before this study was carried out. Genomic DNA was isolated from 0.2 ml of blood with a QIAamp DNA Blood Mini Kit and portions of the hepcidin, HFE, and TFR2 genes were PCR amplified with appropriate primers. Specifically, exons 2, 4, and 6 of the TFR2 gene were amplified. These exons were chosen for analysis as previous studies had shown mutations in these exons [92–94]. The resulting amplicons were heated to 94 °C for 10 min cooled at 56 °C for 60 min to obtain heteroduplexes. Mutations were analyzed by denaturing high-performance liquid chromatography on a Transgenomic WAVE® System with a preheated C18 reversed-phase column. Eight μL of the PCR mixture were loaded into column duplexes were eluted at a flow rate of 0.9 μL/min. DNA was detected by monitoring the absorbance at 260 nm and DNA fragment melting characteristics were predicted by Wavemaker™ software. The samples with altered elution profiles were subjected to DNA sequencing. As this technique does not detect homozygous mutations, the amplicons were also mixed with equal amounts of wild-type DNA, denatured, reannealed, and analyzed for heteroduplexes. All identified C282Y and H63D HFE gene mutations were confirmed by enzymatic digestion. No discrepancies between endonuclease assay- indentified C282Y and H63D HFE gene mutations and denaturing high-performance liquid chromatography HFE gene mutation results were detected, indicating that the chromatographic assay worked well and had 100 % sensitivity, at least for these two mutations.
Out of the 657 individuals tested, three new intronic variations, a previously identified IVS5-9T > A transition, and a new Val22 → ILE TFR2 gene mutation where found. Only one individual (72 years old and hepatitis B surface antigen positive) with TFR2 gene mutations and no concomitant HFE gene mutations had a high serum ferritin value, possibly due to viral hepatitis. Based on these results, the authors concluded that HFE and TFR1 gene mutations are often associated in HH and that HH due solely to the rare TFR2 mutations is uncommon.
7.6.5 SLC40A1 Gene Mutation and Type 4 HH
The SLC40A1 gene mutations cause a rare autosomal dominant HH, showing unusual clinical features including high serum ferritin levels with low or normal transferrin saturation until the end stage of the disease. Additionally, urinary hepcidin is usually reduced. Strangely, it may initially present as mild iron-deficient anemia with a reduced tolerance to therapeutic phlebotomy. SLC40A1 consists of eight exons and encodes the ferroportin protein – a multipass transmembrane protein located on the basolateral membrane of enterocytes that regulates iron export into the circulation [10, 95]. Ferroportin also regulates the efflux of iron from macrophages of the spleen, liver, and bone marrow, and Type 4 HH is unique in that it often shows increased Kupffer cell iron ([10, 95, 96], Fig. 7.3a, b). SLC40A1 gene mutations lead to increased iron loading, often due to ferroportin losing its responsiveness to hepcidin [97].
Like molecular testing for HH types 2 and 3, SLC40A1 gene mutation analysis is not commonly performed, due to the rarity of the disease. Most molecular tests involve sequencing the entire coding region of the gene. Montosi et al. [98] examined genomic DNA isolated from peripheral blood from an Italian pedigree that included 15 individuals with iron overload, resulting in hepatic fibrosis, diabetes, impotence, and cardiac arrhythmias. None of these 15 individuals carried the C282Y HFE gene mutation [99]. The ferroportin exons were PCR amplified, sequenced, and compared to control sequences. Individuals with iron overload, but not those with normal iron levels, showed an exon 3 mutation that converted an alanine at residue 77 to an aspartic acid (GCC → GAC). The authors hypothesized that this mutations resulted in a gain of protein function that resulted in increased dietary iron accumulation. At least 26 other ferroptorin mutations have been identified by various molecular tests [10].
7.6.6 Genetic Testing for HH
While most cases of HH are due to HFE gene mutations in European populations, a subset of HH in this population and non-European populations can carry different gene mutations. Santos et al. [10] recommended the testing algorithm shown in Table 7.1.
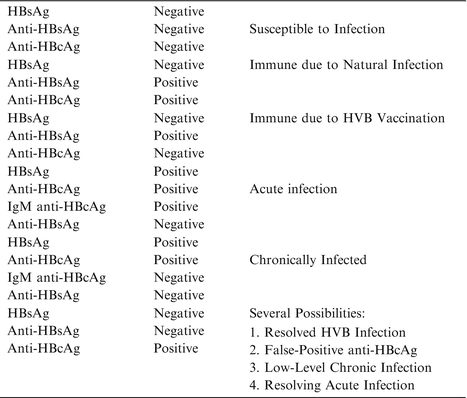
Table 7.1
Common serological patterns associated with HBV infection (105–107)
7.7 Molecular Diagnostic Testing for Hepatitis B and C
Chronic HBV, HCV, and the satellite virus of HBV, hepatitis D (HDV), are major risk factors for HCC, with approximately 500 million people infected with viral hepatitis worldwide [1–4]. Proper medical management of chronic viral hepatitis requires timely and accurate diagnosis, staging, and disease monitoring. In general serological markers are commonly used for viral hepatitis diagnosis and staging, while molecular diagnostic tools are used for the identification of specific viral strains and for assessing drug responses. The end-point of such treatment is obtaining a sustained virological response, preferably with undetectable serum viral RNA, 6 months post-therapy completion [1–4, 99, 100]. Here we will review the serological and molecular testing for HBV, HCV, and HDV, with emphasis on molecular diagnostic techniques.
7.7.1 HBV
The WHO estimates that over two billion people have been infected with HBV and currently some 350 million people are chronically infected worldwide, with about 75 % living in the Asia Pacific region. Chronic HBV confers an increased risk for hepatic decompensation, cirrhosis, and HHC, and it is estimated that the worldwide mortality from this disease is 0.5–1.2 million deaths per year. In adults, most HBV infections are eventually cleared with only about 5 % resulting in chronic infections. In neonates, however, over 90 % fail to clear the virus and become chronically infected. HBV is spread parenterally by percutaneous or permucosal exposure to infected body fluids, especially blood. Risk factors for HBV infection include re-using syringes in between intravenous drug users, transfusions with untested blood, tattooing with unsterilized instruments, sexual promiscuity, working in a health-care setting, and living in a correctional facility [101, 102].
HBV is a double-stranded (ds), circular, enveloped 3,200 base pair DNA virus of the hepadenaviridae family. HBV has at least eight major genotypes (A through H), which shows a greater than 8 % divergence in viral nucleic acid sequence. Following entrance of the HBV into the hepatocyte, the viral genome enters the nucleus and integrates into the host genome, where the viral DNA functions as a transcriptional template making viral RNA. Unfortunately, because HBV integrates into the host genome and remains as an intercellular replicative intermediary (covalently closed circular DNA) complete elimination of HBV from an infected individual is not usually possible. Some of the viral RNA intermediates are reversed transcribed into DNA. The transcription products are used to make viral proteins, including the HBV surface and e antigens. Although HBV has a strong preference for the liver, viral DNA can be found in the kidneys, pancreas, and mononuclear cells of infected individuals. In acute infections the HBV infection becomes apparent roughly 45–180 days post initial infection, with the first 6 months of infections defined as acute phase of infection. Typically clearance of the HBV infection begins several weeks prior to the onset of clinical symptoms, and it is caused by antiviral cytokines produced by the adaptive and innate immune responses. Later, a cytologic immune response causes hepatocyte necrosis and apoptosis, responsible for the concomitant onset of clinical symptoms, and a rise in serum alanine aminotransferase (ALT). Individuals with a strong and specific CD4/CD8 immune response will usually clear the HBV, while a weaker response is associated with chronic HBV [101, 102].
7.7.1.1 HBV Serologic and Histopathologic Testing
Liver damage and function are typically analyzed serum ALT, bilirubin, and albumin levels, and the prothrombin time. Liver biopsy is used to quantify the grade of inflammation and the stage of fibrosis. Non-invasive methods such as ultrasound or rising alpha-fetoprotein levels can, on the other hand, often detect cirrhosis and HCC. Several serologic tests are employed in HBV infection and serum analysis based assays are presently the mainstay testing modality. Detectable HBV surface antigen (HBsAg) is a marker of chronic infection, while viral core antigen (HBcAg) indicates high hepatic viral replication. Loss of serum HBcAg with concomitant raise in anti-HBcAg antibodies (termed seroconversion) is associated with clinical improvement and reduced serum ALT, hepatitis, and reduced HBV DNA levels [101–103]. Serum analysis of HBV-related antigens and antibodies identify the different phases of the infection and are summarized in Table 7.1 [101–103].
7.7.1.2 HBV Molecular Genetic Testing
Molecular HBV testing is usually employed in the diagnosis and management of HBV infection; quantification of viral load, genotyping, drug resistance, mutation analysis, and core promoter/precore mutation assays. Of these assays, the quantification of viral load is the most commonly used test [103].
7.7.1.2.1 HBV Viral Load Quantification
HBV viral load quantification is usually performed on plasma or serum. It is used to measure the viral load prior to the initiation of therapy, to monitor the effectiveness of anti-viral therapy, and to identify “occult” HBV viral infections, where the serologic tests can fail to detect HBV infections. It is also important in liver transplantation to avoid unnecessary HBV transmission [101–103]. In general, individuals with chronic HBV that are HBsAg positive, HBeAg negative, anti-HBeAg positive and serum HBV DNA levels below 105 copies/ml (>20,000 IU/ml) show histologically mild HVB-induced hepatic lesion as graded according to Ishak et al. [104, 105].
7.7.1.2.2 Nucleic Acid Hybridization Assays for HBV Quantification
The first molecular assay employed to quantify HBV viral loads were hybridization assays. Although these assays worked well, they were less sensitive and more cumbersome to perform that most PCR-based assays and are now seldom performed [104]. The degree of HBV probe hybridization does directly quantify the amount of serum HBV DNA present and correlates closely with infectivity [106]. Guo et al. [107] employed digoxigenin-labeled probes to detect HBV DNA in serum. Full-length HBV was inserted into a pBR322 plasmid at a PstI site, grown up in bacteria, subjected to restriction endonuclease digestion and isolated by gel electrophoresis. The probed DNA was purified by ion-exchange chromatography and either labeled with [α-32P]dATP with a random labeling heximer or with a digoxigenin-11-dUTP labeling according to Boehringer Mannheim kit protocol. HBV DNA was isolated from human sera by two means: phenol-chloroform extraction followed by ethanol precipitation, or by alkaline denaturation in 1 M NaOH, followed by neutralization. Both samples were applied to nitrocellulose prewetted with appropriate buffers, baked to attach the HBV DNA to the nitrocellulose, pre-hybridized, hybridized with the HBV probes, washed, dried, and exposed to X-ray film. For the digoxigenin-labeled probes, the labeled DNA was detected immunologically using an antibody-enzyme conjugate, anti-digoxigenin-alkaline phosphatase with an enzyme-linked color reaction. Both assays included a standard curve within the same blot. Both assays yielded a lower detection limit of ~0.25 pg, corresponding to 7.0 × 104 genome copies. Since this number is close to the 105/ml copy number that defines viral loads that are not usually damaging to the liver. The sensitivity of this hybridization assay is adequate, but the sensitivity of the PCR assay is greater. Additionally, the digoxigenin-based assay had a longer shelf life and is safer to work with than the 32P-based assay.
7.7.1.2.3 Branched DNA Technology in HBV Detection
Branched chain DNA (bDNA) is a sensitive, specific, and reliable tool for the diagnosis of viral infections and in monitoring drug response and/or disease progression. It has been successfully on multiple microorganisms, including T. bricei, cytomegalovirus, human papilloma virus, HIV, HCV, and HBV. bDNA does not depend on nucleic amplification, but instead uses a bDNA probe that hybridizes to the target sequence(s), resulting in a complex of probes and target sequences that can be quantified with great accuracy.
To perform this assay HBV is isolated from patient serum and treated to insure that the DNA can bind the assay “extender probe”. The assay is usually performed on a multi-well plate, such as a 96 well plate. Attached to the walls and floor of the plate are many “capture probes”. Extender probes are added under the correct buffer conditions to allow the capture and extender probes to hybridize. Part of the extender probe binds to the capture probe while the other, non-bound, single-stranded portions hybridizes to the target HBV DNA. A “label extender” is then added which binds to contiguous regions of the bound target HBV DNA. A “preamplifer oligonucleotide” is added which hybridizes to the label extender. This oligonucleotide partially hybridizes to the label extender and also usually has multiple branched oligonucleotides that hybridize to an amplifier oligonucleotide having alkaline phosphatase attached to it. After the required multiple washing steps, the amount of HBV target DNA present in the serum sample will be directly related to the signal created by the reporter molecule (alkaline phosphatase). A standard curve with positive and negative controls is used to estimate the number of HBV genomes in the serum sample. Essentially this assay depends on the HBV DNA linking the fixed capture probe to the amplifier/reporter, allowing the amount of HBV DNA to be quantified [108, 109]. The procedure may be summarized as follows:
Well → Capture Probe → Label Extender → HBV Target DNA → Label Extender → Preamplifier Oligonucleotide → Amplifier Oligonucleotide-Alkaline Phosphatase Conjugate
The advantages of bDNA technology include the fact that the technique lacks any amplification steps (therefore lowering the chances of cross contamination), that it is easy to automate, and is not labor intensive. Its sensitivity is usually less than that of PCR-based assays, but its reproducibility and specificity are excellent. Thus this assay would work well in patient populations that typically have higher HBV titers [109].
7.7.1.2.4 HVB Quantification by PCR
PCR has the advantages of high sensitivity and specificity and has largely replaced hybridization assays for serum HBV DNA quantification. While the sensitivity of hybridization assays is limited to about 70,000 viral genomes/ml, many PCR assays can reliably detect serum viral genomes over a 101 and 108/ml range [107, 110]. Abe et al. [110] developed an early RT-PCR assay for quantifying serum HBV. To begin this work, nucleotides 1–2,182 of the HBV genome were inserted into a BlueScript II SK(+) plasmid (Stratagen), grown in bacteria, and the recombinant plasmid was purified and then quantified by the optical density at 260 nm. Real-time detection PCR was performed on the HBV DNA with three sets of PCR primers and a probe; two primer sets would amplify the HBV surface (S) antigen gene and one the viral X gene. The amplified HBV viral gene segments were chosen based on being highly conserved between different viral strains, on having high sensitivity/specificity, and being placed relatively widely apart within the viral genome.
To quantify the HBV load, DNA was extracted from 100 μl of plasma and place into 20 μl of water. 10 μl of this DNA was amplified in a 50 μl volume with one of the primer-probe sets, Taq polymerase, appropriate buffer conditions, and nucleotide triphosphates. The mix was subjected to amplification cycles of 95 °C for 20 s and 60 °C for 1 min. The amount of target DNA was quantified by measuring the increase in probe fluorescence, following probe degradation with concomitant probe-quencher disassociation, normalized to an unchanging passive reference signal. The amount of target DNA amplified was thus the change in probe fluorescence minus the reference signal measured at the beginning of the PCR reaction. The threshold Ct value was calculated on the mean baseline of the first 15 amplification rounds and set at 10 standard deviations above this mean. The amount of HBV target DNA was measured against a standard HBV DNA curve ranging over 101 to 108 copies/reaction. A linear relationship was obtained between the Ct and the number of HBV viral copies. In parallel experiments employing HBV DNA quantification by bDNA technology, the detection limit was found to be 7 × 105 viral copies/ml. When applied to the sera of HBV infected and viral-free individuals, this PCR protocol was able to detect HBV viral DNA over a life range (101–108 copies/reaction) and have similar sensitivity of bDNA technology. This assay was also able to detect HBV DNA in patients who had undetectable HBV level by the bDNA testing method. The authors of this early study conclude that this assay was superior to other methods for HBV viral DNA quantification in its sensitivity, specificity, simplicity, and reproducibility.
Since this study, RT-PCR has become the gold standard for HBV DNA detection in patient sera and this technique is routinely employed in the automated detection of serum HBV [111–113]. However, RT-PCR still has limitations in detecting very low HBV loads. Wang et al. [114] used a locked nucleic acid PCR (LNA-PCR) probe to quantify serum HBV loads compared to standard TaqMan RT-PCR probe and achieved a significant increase in the viral detection limit. LNAs carry 2-methyele units between the O2 and C4 of ribose, locking the ribose in the 3′-endo (North) conformation, often found in A DNA. This locked conformation enhances base stacking and increases the melting temperature 2–8 °C compared to unmodified RNA or DNA oligonucleotides, increasing mismatch discrimination [115]. Wang et al. [114] used an LNA or an unmodified oligonucleotide probe designed to match the HBV X gene and produce a 114 base pair amplicon. The same oligonucleotide primers were used in both reactions:
Primer 1 | GACCACCAAATGCCCCTAT |
Primer 2 | CCRAGAYYGAGATCTTCTGCGAC |
LNA probe | FAM-TCGTCTAACAACAGT-BHQ1 (underlines denote methylene bridge) |
TaqMan probe | FAM-TCGTCTAACAACAGT(TAMMRA)AGTTTCCGGAAGTGT-P |
Serum samples were taken from 39 individuals, previously demonstrated by have chronic HBV by ELISA. These samples were divided into three groups: (1) 15 cases that were HBsAg positive only, (2) 10 cases that were HBsAg and HBeAg positive, and (3) 14 cases that were HBsAg, HBeAg, and HBc positive. Additionally, 19 cases of normal serum were taken from volunteers. The HBV DNA was isolated from 100 μl serum and 2 μl of purified DNA were used per reaction out of a final 25 μl volume. The amplification reaction consisted of 40 μl of 200 nM each primer, 75 nM LNA probe, dGTP, dATP, dCTP, and dUTP, 3.5 mM MgCl2, 2 U HotSartTaq DNA polymerase, 0.5 U uracil DNA glycosylase and, 2 μl of purified DNA. The amplification was carried out in a iCycler iQ5 (Bio-Rad) consisting of an initial activation of UDG at 37 °C for 5 min, activation of HotStarTaq DNA polymerase and template denaturation at 95 °C for 3 min, and 40 cycles in two steps: 95 °C for 5 s, 60 °C for 30 s. Standard curves were created in the range of 40–4 × 107 IU/ml by 1:10 serial dilutions of an HBV standard. All samples were run in duplicate.
A linear relationship was obtained between the Ct values and the log10 concentrations of HBV DNA. Over a serial dilution of 40–4 × 107 IU/ml the maximum sensitivity of the standard oligonucleotide primers was 80 IU/ml, while the maximum sensitivity for the LNA probe was 40 IU/ml. Thus the LNS probe roughly doubled detection sensitivity compared to the standard oligonucleotide probe. When applied to patient samples, both probe types were able to detect HBV DNA in patient samples. Interestingly, in the 15 HBsAg positive only samples with low HBV DNA, the LNA probes were able to detect HBV DNA in four samples determined to be HBV DNA negative by standard probes. The authors concluded that LNA probe has higher precision, an improved signal to noise ration, and a better detection limit in clinical assays compared to standard probes.
7.7.1.2.5 HBV Molecular Genotyping Assays
HBV has at least eight different genotypes, A–H, which show greater than 8 % genomic variation between the viral types and numerous subtypes which show at least 4 % genomic variation. Most likely these variations have arisen due to the relatively high rate of nucleotide replication substitution (1.4–4.57 × 10−5 per base pair/year) seen in HBV. Phylogenetic evidence suggests that human HBV is a relatively recent viral pathogen, that first appeared about 3,000 years ago [116, 117]. Many of the HBV genotypes show significant differences in: (1) geographic distribution, (2) clinical course, (3) treatment responses, and (4) modes of transmission. For example, HBV genotype A is more common in Northwest Europe and the USA, while HBV genotype C is more common in Asia and the Pacific region. The HBV genotype C induces a more severe disease, characterized by higher fibrosis scores and a more frequent progression to cirrhosis than does HBV genotype B, while the D genotype appears to cause a higher incidence of HCC [118–122]. Additionally, while all HBV genotypes appear to respond equally well to nucleoside/nucleotide analogue treatment, genotypes A and B respond better to pegylated interferon-α than do genotypes C and D following 52 weeks of treatment [123, 124]. Last, different HBV genotypes are preferentially spread by different modes of transmission, although all genotypes can be spread by the common transmission modes. For example, HBV genotype A has been associated with sexual transmission while the D genotype is often transmitted by blood transfusions and IV drug abuse. Interestingly, genotype A appears to also be preferentially transmitted by homosexual or bisexual contact [125, 126].
Presently there are roughly ten different HBV genotyping techniques that entirely or partially use molecular technology (for review, [127]). Here these techniques will be briefly reviewed and the advantages and disadvantages of each technique discussed.
7.7.1.2.6 Direct HBV Sequencing
Sequencing remains the gold standard for HBV genotyping and presently commercial kits are available for this purpose, such as HBV sequencing test kit offered by Abbott Molecular (Illinois, USA). Sequencing has the advantage of directly interrogating every nucleotide within the viral genome or viral gene being sequenced, allowing the identification of new sequence variations, and it can be a relatively sensitive test. Since all nucleotides are interrogated, sequencing works equally well for genotyping, identifying drug resistant HBV clones, and other mutation testing. The drawback of this technique is that it can be expensive, time-consuming, and can easily miss less commonly represented HBV genotypes within a genotype mix [127].
Okamoto et al. [117] developed an early method for Sanger sequencing of the HBV genome. HBV DNA was isolated from 20 ml of serum taken from individuals demonstrated to have HBV by solid phase immunoassay for HBsAg and by dot blot hybridization for HBV DNA. The gapped region of the HBV genome was repaired with an endogenous DNA polymerase and the HBV DNA was cloned into a pSP65 plasmid BamHI, grown in bacteria, and subcloned into different phage vectors. The HBV genomic fragments were removed with the appropriate restriction endonuclease and subjected to standard Sanger sequencing. A total of 18 HBV genomes were sequenced, five of which comprised newly identified viral subtypes. Although an early study, this work did demonstrate that Sanger sequencing is an excellent, although slow way to analyze HBV viral sequence variations.
Since the eight different HBV genotypes show significant sequence variation, efficient and accurate genotyping methods employing Sanger sequencing combined with genotype-specific PCR primers have been developed. The commercial TRUGENE HBV Genotyping commercial kit (Siemens Medical Solutions Diagnostics, NY, USA, [128]) uses these techniques. First, DNA is extracted from serum and the HBsAg gene is amplified via PCR. The resulting amplicons are then bidirectionally Sanger sequenced and compared to the known A through H HBV genotypes via a bioinformatics program. The bidirectional Sanger sequencing improves accuracy, as each sequence is effectively read twice. The detection limit of this assay is approximately 2.0 × 103 HBV genome copies/ml. Although less commonly used, pyrosequencing has worked well for some HBV sequencing applications, such as the analysis of specific nucleotide substitutions associated with dug resistance [129].
7.7.1.2.7 Multiplex Polymerase Chain Reaction
Multiplex PCR has been used to analyze HBV genotypes A through H in an accurate and cost-effective manner. Liu et al. [130] employed this technique to analyze the HBV genotypes in the sera of 441 HBV DNA-positive patients and compare it to restriction fragment length polymorphism analysis. The primers employed in this study were designed by analyzing 369 HBV full-length nucleotide sequences in GenBank which had been classified into eight different genotypes (A–H) using the Clustal X1.81, GenDoc2.6.002 and Mega2 software. From this database, genotype-specific primers were designed that were specific to at least 90 % of the viral isolates corresponding to the genotype.
Following patient DNA isolation, the HBV DNA was amplified by multiplex PCR performed with a GeneAmp PCY System 9600 in a 50 ml volume. The amplification protocol incubated the samples for 5 min at 94°, followed by 40 cycles consisting of 94° for 1 min, 59.5° for 1 min, and 72° for 2 min. The amplicons were visualized by electrophoresis in a 4 % agarose gel, stained with ethidium bromide, and evaluated under UV light. Each primer set gave an amplicon with a different molecular weight, allowing identification of the HBV genotype. HBV infections containing different genotypes gave multiple bands of different sizes. In order to be detected, a co-infecting HBV genotype had to represent at least 10 % of the total HBV infection load. When the results of the multiplex PCR were compared to genotyping results obtained by restriction fragment length polymorphism (RFLP) analysis a 93 % concordance was achieved, indicating that genotyping with multiplex PCR is relatively accurate. The authors concluded that this genotyping method could be applied only to areas where HBV genotypes A–G are prevalent. It could also provide an efficient alternative for clinical diagnosis and large-scale studies due to its low cost and fast turn-around time. PCR has also been combined with melting curve analysis to identify different HBV genotypes. This technique allowed as little as 100 HBV genome copies/ml sera and was more accurate than RFLP analysis [131].
7.7.1.2.8 Restriction Fragment Length Polymorphism (RFLP) Analysis
RFLP analysis interrogates the sequence difference between to homologous DNA sequences by using restriction endonucleases to cut the DNA into fragments of different lengths based on the sequences having sites cut by different restriction endonucleases. The resulting DNA fragments are often resolved by gel electrophoresis. Lindh et al. [132] used RFLP to genotype HBV. Seventy-three HBV sequences were obtained through the Entrez database of the National Center for Biotechnology Information (NCBI, Bethesda, MD) and analyzed with MacVector DNA analysis software (International Biotechnologies, New Haven, CT). A restriction endonuclease fingerprint pattern was predicted following treatment with the Tsp509I, HinfI, and BsrI endonucleases. To test the predicted fingerprint pattern, 187 HBeAg-positive chronic HBV carriers who resided in western Sweden were then analyzed by a commercial HBV genotyping serology kit (Abbott, Abbott Park, IL), or by RFLP. To perform the RFLP DNA was prepared from 30 μl of patient serum and the segment between nucleotides 256 and 796 were PCR amplified by 40 PCR cycles. Three minutes denaturation at 94 °C, 40 cycles of amplification, including denaturation for 45 s at 94 °C, annealing for 60 s at 53 °C, and extension for 90 s at 72 °C (prolonged by 3 s/cycle), were done and followed by a final 7 min extension at 72 °C.
The resulting amplicon was incubated for 3 h with either the Tsp509I or the HinfI restriction endonucleases. The cut amplicons were then run into a composite gel containing 2 % NuSieve agarose and 1 % standard agarose, with uncut amplicon as a negative control. In some cases the cut amplicons were cut further with the BsrI endonuclease. The DNA fragments were identified with ethidium bromide staining and the restriction fragment pattern read visually. One hundred and sixty-six of the samples fell into one of ten major patterns, each corresponding to specific HBV genotype. Seven additional patterns appeared to represent rare HBV genotypes. Thus roughly 92 % of the HBV genotypes were easily identified. Of the others, 14 samples were incubated with BsrI, allowing seven additional cases to be genotyped. Comparison of this method to serological testing showed a 100 % genotyping correlation for those samples that could be RFLP analyzed.
Related posts:
 Molecular-Genetic Testing in Penile, Scrotal, and Testicular Cancer
Molecular Diagnostics of Pancreatic Cancer
Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Cutaneous Malignancy
Molecular Diagnosis of Bladder and Kidney Cancer
Molecular Pathology and Diagnostics in Esophago-gastric Cancer
Molecular-Genetic Testing in Penile, Scrotal, and Testicular Cancer
Molecular Diagnostics of Pancreatic Cancer
Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Cutaneous Malignancy
Molecular Diagnosis of Bladder and Kidney Cancer
Molecular Pathology and Diagnostics in Esophago-gastric Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


