Two recent studies on whole exome/genome sequencing of 87 and 88 human HCCs as well as matched normal tissue46,47 confirmed the previous studies that β-catenin (10% and 15.9%) and TP53 (18% and 35.2%) are the most frequently mutated oncogene and tumor suppressor, respectively, in HCC.46,47 The study by Kan et al.47 also detected several drugable mutations, including activating mutations of Janus kinase 1 (9.1%), which might provide an option for novel individualized therapeutic interventions. Interestingly, Nault et al.,48 using traditional Sanger sequencing, identified somatic mutations activating telomerase reverse transcriptase as both the earliest and the most frequent mutations in human preneoplastic lesions (25%) as well as hepatocellular carcinomas (59%) and is associated with activating CTNNB1 mutations.48
Compared to whole-genome sequencing, the application of RNA sequencing in liver cancer is currently limited to three studies. One study investigated the transcriptomes of 10 matched HBV-related HCC cases, identifying a total of 1,378 differentially expressed genes with a specific enrichment of chromosome location on 8q21.3 to 24.3.49 Another study investigated three paired nontumor and tumor specimens demonstrating that ADAR1-mediated AZIN1 RNA editing is linked to tumor initiation and development in liver cancer.50
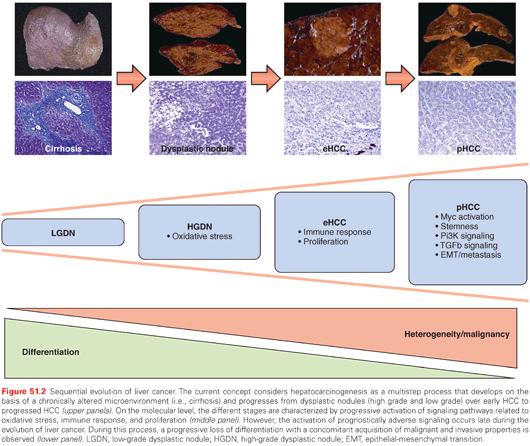
Sequential molecular alterations during human hepatocarcinogenesis from dysplastic lesions to eHCC and, ultimately, pHCC are not clearly defined. This lack of information represents a major challenge in the clinical management of patients at risk. Although recent results associating MYC activation with early stages of malignant conversion into HCC, detailed molecular sequences that drive premalignant lesions into pHCC still remain to be clarified.51 In the third study, integrative transcriptome sequencing to the tumor-free surrounding liver (n = 7), low-grade (n = 4) and high-grade dysplastic lesions (n = 9), eHCC (n = 5), and pHCC (n = 3) from eight HCC patients with HBV infections was applied.52 The results of the study indicate that molecular profiles of dysplastic lesions and eHCC are quite uniform. In contrast, a sharp increase in heterogeneity on both mRNA and DNA levels is observed in progressed HCC. These molecular alterations result in massive deregulation of key oncogenic molecules such as transforming growth factor beta 1 (TGF-β1), MYC, PI3K/AKT, and suggest that activation of prognostically adverse signaling pathways is a late event during hepatocarcinogenesis (Fig. 51.2).
THE MICROENVIRONMENT OF LIVER CANCER
HCC develops on the basis of chronic liver disease and, in more than 80% of the cases, with preexisting liver cirrhosis. For a complete understanding of the molecular mechanisms of hepatocarcinogenesis, the underlying liver disease leading to a chronically altered inflamed liver microenvironment has to be appreciated.53 Recent research efforts have focused on the identification of key factors that contribute to the disruption of the liver microenvironment and the generation of an adverse niche(s) that promotes hepatocarcinogenesis. Among the most prominent factors involved in the so called inflammation-fibrosis-cancer axis is the nuclear factor kappa B (NF-κB) pathway.54 The dominant role of this pathway for hepatocarcinogenesis is well documented.54 However, the absence of NF-κB by genetic loss of the NF-κB master regulator NEMO significantly enhanced liver cancer development in a mouse model, indicating that inhibition of NF-κB may not only exert beneficial effects, but also may negatively impact hepatocyte viability, especially when NF-κB inhibition is pronounced.55
The importance of the microenvironment is further highlighted in a recent study demonstrating that transplantation of hepatic progenitor cells gave rise to cancer only when introduced into a liver with chronic damage and compensatory proliferation.56 Interestingly, similar to observations made in human hepatocarcinogenesis, the cells resembling these progenitor cells quiescently resided within dysplastic lesions for several months before the appearance of HCC. During this time, the progenitor cells acquired autocrine interleukin (IL)-6 signaling that stimulated in vivo growth and malignant progression, which might be a general mechanism of progenitor cell–induced HCC.
However, the microenvironment does not only contribute to tumor initiation. Although gene-expression profiles of tumor tissue failed to yield a significant association with survival, a 186-gene signature generated from the surrounding nontumoral liver tissue was highly correlated with outcomes in a cohort of more than 300 HCC patients.57 Consistently, this poor-prognosis signature contained gene sets associated with inflammation, such as IFN signaling, NF-κB, and tumor necrosis factor α (TNF–α). Further, a gene set enrichment analysis showed that the downstream targets of IL-6 were strongly associated with the poor-prognosis signature, again confirming the importance of this signaling for hepatocarcinogenesis.
Together, these studies demonstrate the complexity of molecular mechanisms influencing the development and progression of liver cancer that are exerted by epigenetic and genetic alterations and a cross-talk between microenvironment and damaged hepatocytes and/or cancer cells, respectively.
CLASSIFICATION AND PROGNOSTIC PREDICTION OF HEPATOCELLULAR CARCINOMA
The application of microarray technologies to characterize tumors on the basis of global gene expression has had a significant impact on both basic and clinical oncology.10 The goal of tumor microarray studies generally includes the discovery of subsets of tumors (class discovery), which enables diagnostic classification (class comparison), a prediction of clinical outcome (class prediction), and mechanistic analysis. Verification and validation of the primary results are essential for the discovery of oncogenic pathways and the identification of therapeutic targets.8
The goal of all staging systems is to separate patients into homogeneous prognostic groups to permit the selection of the most appropriate surveillance as well as to find a specific therapy for each subtype. Although much work has been devoted to establishing prognostic models for HCC by using clinical information and pathologic classification, many issues still remain unresolved.58 More than 20 studies on prognostic HCC gene expression profiling, as well as several reviews, have appeared during the last 10 years.4 However, results from these studies are quite heterogeneous and, besides disruption in general cancer-related processes such as proliferation, apoptosis, neoangiogenesis as well as prometastatic and proinflammatory gene sets, the overall similarity was low, thus limiting a successful implementation into clinical practice. A potential explanation is the fact that the interpretation of molecular profiling studies of HCC poses more challenges than other human tumors, mainly because of the complex pathogenesis of this cancer.59As already emphasized, HCC arises in diverse settings ranging from infection with HBV or HCV to chronic metabolic diseases as varied as diabetes, NAFLD, and hemochromatosis. These different disease stages represent complex assortments of genetic and epigenetic aberrations as well as altered molecular pathways.39,60
A recent study aimed to generate a composite prognostic model by evaluating 22 prognostic gene expression signatures generated from tumors as well as cirrhotic tissues in a cohort of 287 patients with early stage HCC (BCLC 0/A).61 Overall, most previously reported signatures retained their prognostic ability in this independent data set. Out of these 22 signatures, 17 were able to adequately subclassify patients according to their prognostic trait. It is noteworthy that none of the signatures reflecting a progenitor cell origin (i.e., EpCAM, hepatoblastoma-C2, CK19-rat, CK19-human signature) could be confirmed to be of prognostic value. However, these signatures have not been generated for the classification of early stages of HCC.
Another important finding of this study was the observation that gene expression profiles obtained from paired biopsies from the center and periphery of the same tumor in 15 tumor specimens showed a high (>80%) transcriptomic concordance. Although these observation provide at least some evidence for stability of gene expression signatures in paired biopsies and suggests a low influence of sampling error, more in-depth analyses are needed to better define the intratumoral genetic heterogeneity of HCC that is likely to contribute to the high tumor recurrence and chemoresistance of the disease.62,63
Nault et al.64 aimed to identifying a robust molecular signature to accurately predict the clinical outcome of HCC patients who underwent curative surgical resection. They identified a panel of five genes (TAF9, RAMP3, HN1, KRT19, and RAN) in a training cohort of 314 HCC patients with a strong prognostic relevance and further validated this panel in two independent validation cohorts with different HCC etiologies.64 The five-genes panel was associated with disease-specific survival both in the training and in the validation cohort, and was found to be significantly more accurate in predicting the patients’ prognosis (i.e., survival and recurrence) when compared to known gene expression signatures. Due to the simplicity of the panel and its reproducibility in different technologies (i.e., gene expression microarray and real-time reverse-transcription polymerase chain reaction [qRT-PCR]), the work by Nault et al. has a high potential for implementation into the clinical management of HCC. In particular, the score may be helpful to stratify patients at high risk for relapse and tumor-related death before the decision of liver resection or transplantation is made. However, independent validation of this prognostic algorithm by different groups and independent cohorts is needed before the panel can be used for clinical decision making.
Two independent studies provide evidence that the oncofetal marker SALL4 could be an attractive therapeutic target in HCC with progenitor cell origin.65,66 High SALL4 expression was significantly associated with overall survival of patients in large independent cohorts. Further, and consistent with its oncofetal role, SALL4 overexpressing tumors shared similar molecular profiles with fetal progenitor cells and stemness-associated HCC. Finally, by using RNAi and a specific inhibitory 12-AA peptide against SALL4, the authors could convincingly show that SALL4 possessed therapeutic potential in HCC by interacting with PTEN/PI3K-AKT signaling via the interaction with the NuRD complex.67
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


