The mutation spectrum (Fig. 38.2B) differs significantly from that found in other cancers. Mutations E542K and E545K in the helical domain are most common (22% and 60%, respectively) and the kinase domain mutation H1047R, which is the most common mutation in other cancers, is less frequent. The selective pressure for helical domain mutation in UC is not fully understood. E542K and E545K forms require interaction with RAS-GTP but not binding to p85, the regulatory subunit of PI3K, whereas H1047R depends on p85 binding and is active in the absence of RAS binding.29 This suggests potential cooperation between helical domain PIK3CA mutant proteins and events in UC that activate RAS. Compatible with this, PIK3CA and FGFR3 mutations are commonly found together.25,26,27,28 Mutant PIK3CA confers a proliferative advantage at confluence and stimulates intraepithelial movement in NHUC, with higher activity of helical domain than kinase domain mutants in this cell context.30 Several other mechanisms of PI3K pathway activation have been identified in UC, though none of these are common in noninvasive tumors (Table 38.1).
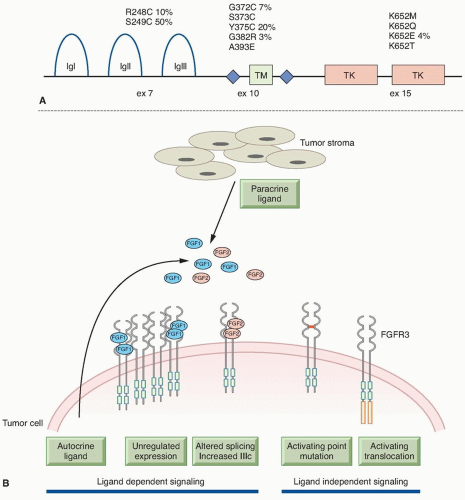 Figure 38.1 (A) FGFR3 mutations identified in bladder cancer. Positions of hot-spot mutations in exons 7, 10, and 15 that are found in bladder cancer are shown in relation to protein structure. The relative frequency of the more common mutations is given as a percentage. IgI, IgII, IgIII, immunoglobulinlike domains; TM, transmembrane domain; TK, tyrosine-kinase domain. (B) Mechanisms of FGFR3 activation in bladder cancer. FGFR3 can be activated by ligand-dependent and -independent mechanisms. Ligand-dependent activation may be via increased expression of wild-type FGFR3, increased production of FGFs by tumor or stromal cells, with or without upregulated FGFR3 expression, or through expression of splice variants with the ability to bind a wider range of FGF ligands. Ligand independent activation can be achieved by point mutation that facilitates receptor dimerization or by the generation of fusion proteins that constitutively dimerise. |
cohesion between sister chromatids following DNA replication to ensure correct chromosomal segregation. Mutations in STAG2 in glioblastoma have been reported to cause aneuploidy35 but this relationship is not apparent in UC, where most mutations have been found in low grade/stage, genomically stable tumors, and there is no association of mutation with chromosomal copy number changes.31,34
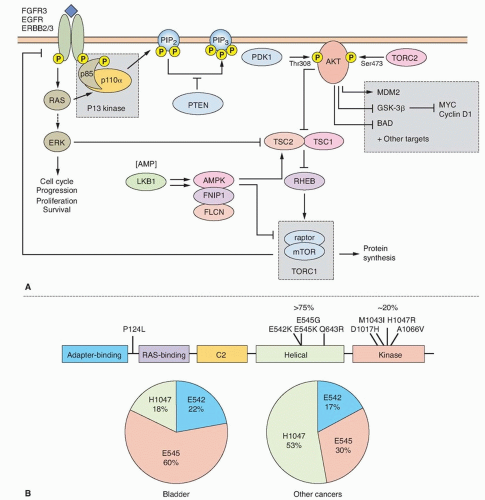 Figure 38.2 (A) Oncogenic signaling via the RAS-MAPK and PI3K pathways. Growth factor-mediated signaling or mutational activation of RAS oncogenes can activate both of these pathways. Signaling via the RAS/RAF/MEK/ERK cascade leads to phosphorylation of many substrates that can have multiple cellular effects depending on the intensity and duration of signaling. In many situations, proliferation is induced. Activated receptor tyrosine kinases bind p85, the regulatory subunit of PI3K, and recruit the enzyme to the membrane where it phosphorylates phosphatidyinositol-4, 5-bisphosphate (PIP2) to generate PIP3, which in turn recruits PDK1 and AKT to the membrane, where AKT is activated by phosphorylation to regulate a wide range of target proteins (not all shown). Among these are cyclin D1 and MDM2, which are upregulated either directly or indirectly, resulting in a positive stimulus via the RB or p53 pathways, respectively. AKT also phosphorylates and inactivates tuberin the TSC2 gene product, leading to activation of mTOR complex 1 (TORC1), which controls protein synthesis. The TSC1 product hamartin forms an active complex with tuberin, and loss of function of either protein leads to dysregulated mTOR signaling. MYC expression is induced as a consequence of both by ERK and AKT signaling. Many genes in these pathways show mutation [FGFR3, PIK3R1 (p85α), PIK3CA (p110α), HRAS, KRAS2, PTEN, AKT1, TSC1, TSC2] or upregulated expression (EGFR, ERRB2, ERRB3, FGFR1) in bladder cancer. (B) PIK3CA mutations identified in bladder cancer in relation to protein structure. Pie charts show proportions of common helical domain (E542, E545) and kinase domain (H1047) mutations in bladder and other cancers. Data from COSMIC (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/; accessed November 15, 2013). |
TABLE 38.1 Genetic Changes Identified in Stage Ta Bladder Tumors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Inactivation of this locus in UC is commonly by homozygous deletion (HD). LOH of 9p, HD of CDKN2A, and loss of expression of p16 in NMI UC is predictive of reduced recurrence-free interval.50,51,52 Mouse knockout and in vitro experiments indicate that p16 and/or p14ARF may be haploinsufficient.53,54 In human UC, this may affect the biology of approximately 45% of cases that have LOH or reduced copy number of 9p21.
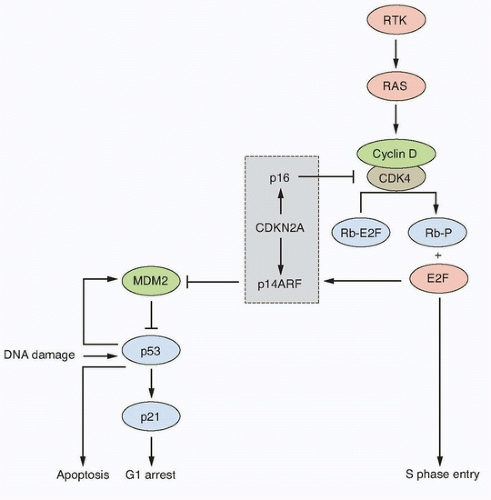 Figure 38.3 Key interactions in the RB and p53 pathways. The CDKN2A locus encodes p16 and p14ARF that act as negative regulators of the RB and p53 pathways, respectively. This interrelated signaling network is central to tumor suppression via the mechanisms of cell-cycle arrest and apoptosis. Stimulation by mitogens induces cyclin D1 expression. Phosphorylation of RB1 by CDK4-cyclin D1 complexes releases E2F family members to induce expression of genes required for progression into S phase. The cyclin D-CDK4 complexes also sequester p27 and p21 (not shown). This allows formation of cyclin E-CDK2, which reinforces the inactivation of RB1. p16 negatively regulates this process by interacting with CDK4. The p53 pathway responds to stress signals (e.g., DNA damage). p21 expression is induced and this leads to cell-cycle arrest. MDM2 is a ubiquitin ligase responsible for inactivation of p53. In turn, p53 regulates MDM2 expression providing a negative feedback loop. The p53 and RB pathways are connected by p14ARF, which sequesters (inactivates) MDM2 in the nucleus and is upregulated by E2Fs and in response to mitogenic signaling. Overexpression of E2Fs and oncogenes such as MYC can both result in p53-triggered cell-cycle arrest via p14ARF. RTK; receptor tyrosine kinase. |
UC cells with epithelial phenotype have enhanced sensitivity to FGFR inhibition compared to those with mesenchymal phenotype.80 A recent RNAi screen in UC cell lines indicated that upregulated EGFR signaling provides a mechanism of escape from FGFR inhibition and can mediate de novo resistance. A reciprocal relationship was found when EGFR was inhibited and both in vitro and in a preclinical in vivo model, combined inhibition showed improved anti-tumor activity.81 Thus, assessment of both FGFR3 and EGFR status may be required to predict response and combined EGFR and FGFR3 inhibition may be essential. Several clinical trials of FGFR inhibitors are now planned or in progress in advanced UC.
TABLE 38.2 Genetic Changes Identified in Invasive (Stage ≥T2) Bladder Tumors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
prognostic significance of FGFR1 upregulation, and FGFR1 has not yet been examined independently of FGFR3 as a potential therapeutic target.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


