SCLC
NSCLC
Frequency
~20–25 %
~75–80 %
Histology
Scant cytoplasm, small hyperchromatic nuclei, fine chromatin, indistinct nucleoli, tumor in sheets
Abundant cytoplasm, pleomorphic nuclei, coarse chromatin, prominent nucleoli, squamous/glandular architecture
Neuroendocrine phenotype
~100 %
Large cell neuroendocrine carcinomas and carcinoids
Peptide secretion
ACTH, AVP, calcitonin, ANF
PTH
Radiation-sensitivity
80–90 % Shrinkage
30–50 % Shrinkage
Chemo-sensitivity
High
Low
Oncogenes
RAS mutation
<1 %
~15–20 %
MYC amplification/overexpression
~15–30 %
~5–10 %
ERBB2 overexpression
<10 %
~30 %
EGFR mutation
Rare
~10 % (Higher in Asians, nonsmokers, adenocarcinomas, females)
EML4/ALK fusion
~5 %
ROS1 fusion
~1–2 %
LKB1 mutation
~9-
HER2 mutation/amplification
~2–4 %
PIK3CA mutation/amplification
~2–18 %
TTF1 amplification
~15 %
BRAF
~2–3 %
MET mutation/amplification
? Rare
~1–20 %
FGFR1 amplification
~22 % SCC
SOX2 amplification
~23 %
Putative autocrine loops
GRP/GRP receptor, SCF/KIT
HGF/MET, neuregulin/ERBB
Tumor suppressor genes
CDKN2A mutation
<1 %
~10–40 %
TP53 mutation
~75–100 %
~50 %
17p LOH
~80–90 %
~70 %
Abnormal TP53 expression (immunohistochemistry)
~40–70 %
~40–60 %
Absent RB1 expression
~90 %
~15–30 %
13q LOH
~75 %
~40–60 %
3p allele loss
>90 %
~50–80 %
9p LOH
~20–50 %
~50–75 %
Absent CDKN2A expression (immunohistochemistry)
~0–10 %
~30–70 %
Other genetic deletions, e.g., 5q, 8p, 11p, 18q
Variable
Variable
Telomerase expression
~100 %
80–85 %
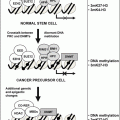
Although it is still uncertain whether SCLC and NSCLC are derived from the same or different cell lineage, much progress has been made towards understanding the molecular basis of lung cancer, particularly involving classic oncogenes and tumor suppressor genes (TSGs or recessive oncogenes). The current paradigm suggests that human epithelial tumors, such as lung cancer, arise as a result of the accumulation of multiple independent molecular events that target critical genetic pathways in key cells. These events appear distinct from the random background genetic damage that is often seen in advanced neoplasms. Specific oncogenes and tumor suppressor genes (TSGs) are the likely targets of somatic aberrations resulting from the genotoxicity of tobacco smoke carcinogens.
The critical cellular pathways affected directly or indirectly by these somatically acquired aberrations are becoming increasingly recognized as biochemical functions of the proteins encoded by mutated genes are unraveled. Cancer is not only caused by abnormal cell proliferation with loss of the usual cellular growth control mechanisms but is also influenced by abnormalities in apoptosis (programmed cell death). Indeed, solid tumors ensue from a balance between cell proliferation and cell death. The regulation of these processes is complex. There are positive factors such as cytokines, hormones, growth factors, and their specific receptors which, upon ligand binding, then signal effector genes via various signal transduction cascades, as well as negative regulators of cell growth and proliferation.
Karyotypic and molecular analyses have demonstrated that lung cancer cells have accumulated several genetic lesions, with perhaps ten or more such events required for the development of an overt lung cancer. Knowledge of the temporal sequence and timing of these genetic lesions in the multistep process of lung carcinogenesis is increasing with improvements in techniques allowing examination of DNA from tiny preneoplastic bronchial lesions, such as with laser capture microdissection. Nonetheless, there appears considerable inter-individual heterogeneity in the timing and number of genetic lesions that occur during lung carcinogenesis; with various alternate pathways leading to bronchogenic carcinoma. While some of the molecular events are commonly found in both SCLC and NSCLC, others show greater specificity for one or other of the subtypes (Table 28.1). These events can largely be categorized into the original and emerging hallmarks of cancer: self-sufficiency in growth signals, insensitivity to antigrowth signals, evasion of apoptosis (programmed cell death), limitless replicative potential, sustained angiogenesis, tissue invasion and metastasis, reprogramming of energy metabolism, and evasion of immune destruction [5].
28.2 Molecular Changes in Overt Lung Cancers
28.2.1 Aneuploidy and Copy Number Variations
Aneuploidy , an abnormal number of chromosomes representing an abnormal total DNA content, is another common characteristic of tumor cells. In solid tumors, aneuploidy is linked to genomic instability, which includes chromosome instability (CIN). With the onset of genomic studies came reports of gene expression signatures associated with aneuploidy and cell p roliferation. Many solid tumors, including lung cancers, often have marked aneuploidy, ranging from hypoploidy to hyperploidy [6, 7]. Several studies have assessed the value of aneuploidy as a marker of biological aggressiveness, but the prognostic value of aneuploidy in lung cancer is controversial [8, 9]. Newer techniques have allowed the identification of extensive areas of aneuploidy in the respiratory epithelium of lung cancer patients, a finding consistent with the field cancerization theory [10].
Early pivotal cytogenetic studies in tumors demonstrated karyotypic features of double-minute chromosomes (DMs) and novel staining regions called homogenously staining regions (HSRs), corresponding to amplified DNA sequences which may contain between twenty and several hundred copies of a specific chromosomal sequence. As the amplified sequences often involve several hundred thousand base pairs, it is possible that more than one oncogene is contained within the amplified region. Chromosomal comparative genomic hybridization (CGH) and array CGH are molecular cytogenetic techniques that can detect gains and losses of DNA in a tumor genome. CGH techniques led to the identification of several new chromosomal regions affected by either deletions or increased DNA copy number in lung cancer genomes [11–15] and include specific allelic loss at 3p, 4q, 9p, and 17p and gain at 1q, 3q, 5p, and 17q [16–20]. More recently, array CGH was developed for high-throughput, high-resolution DNA dosage analysis, where microarrays are spotted with unique genomic probes. Early array CGH microarrays consisted of large genomic clones, such as bacterial or P1 artificial chromosomes (BACs and PACs) or cosmids, robotically spotted onto glass slides, whereas later platforms used short, 25–70-mer oligonucleotides. Moreover, bioinformatic advances have also facilitated the use of single nucleotide polymorphism (SNP) arrays initially developed for genotyping to be used to detect DNA copy number variations [21]. A number of studies have reported microarray-based CGH analyses to detect genomic aberrations in lung cancers, but few in primary SCLC because of its rare resectability (Table 28.2).
Table 28.2
Published array CGH in lung cancer showing copy number variations (DNA gains and losses)
Study | Platform (probes)a | Resolution (kb) | Samplesb,c |
|---|---|---|---|
NSCLC | |||
[23] | BAC (348) | 37 NSCLC (21 SCC, 16AC) | |
[24] | SNP (1494) | 33 Lung cancer cell lines (19 NSCLC, 14 SCLC) | |
[25] | cDNA (8000) | 376 | 14 NSCLC (8AC, 6 SCC), 14 NL |
[26] | BAC (3014) | 1000 | 50 NSCLC (29 SCC, 21AC) |
[27] | cDNA (~10,000) | 1000 | 20 NSCLC cell lines (11AC, 7 SCC, 2 LCC), 9 SCLC cell lines |
[28] | BAC (800) | 27 NSCLC cell lines (11 SCC, 10AC, 6 LCC) | |
[29] | Oligo (22,500) | 54.8 | 44 NSCLC (18AC, 26 SCC) |
[30] | cDNA (12,814) | 8 NSCLC | |
[31] | SNP (115,593) | 51 NSCLC (37AC, 10 SCC, 4 BrAC); 26 NSCLC cell lines (14AC, 4 SCC, 4 LCC, 2 NSCLC, 2 BrAC, 1 AdSq); 19 SCLC tissues; 5 SCLC cell lines | |
[32] | BAC (32,433) | <1000 | 28 NSCLC cell lines (18AC, 9 SCC, 1 LCC) |
[33] | BAC/PAC (4523) | ~700 | 20 NSCLC cell lines (7 SCC, 7AC, 6 LCC) |
[34] | cDNA (12,814) | 20 (8AC, 6 SCC, 5 LCC, 1 AdSq) | |
[35] | cDNA (11,367) | 564 | 23 NSCLC (7AC, 15 SCC, 1 AdSq); 3 metastases; 10 NL |
[36] | BAC | 32 NSCLC | |
[37] | BAC (4046) | 1000 | 36 NSCLC (22 SCC, 14AC) |
[38] | cDNA (~22,000) | ~30 | 76 NSCLC (40 SCC, 36AC), 56 NSCLC cell lines |
[39] | BAC (2464) | 75 NSCLC (43AC, 32 SCC) (Current, former and never smokers) | |
[40] | BAC (4030) | 1000 | 24 NSCLC (12AC, 12 SCC) (with PBMC) |
[41] | BAC (32,000) | 85 NSCLC (56AC, 20 LCC, 9 Other) | |
[42] | BAC (>26,000) | <1000 | 1AC, 1 SCLC, 1 LCNEC (from same patient) |
[43] | BAC (>26,000) | <1000 | 112 LC cell lines (77 NSCLC, 32 SCLC) (with and without EGFR TKI treatment) |
[44] | Oligo (244,000) | 2.1 | 42 NSCLC (from 20 patients: 6 synchronous, 14 metachronous) |
[45] | BAC (4046) | 1000 | 36 NSCLC (22 SCC, 14AC) [37] |
[46] | 76 NSCLC (40 SCC, 36AC), 56 NSCLC cell lines [38] | ||
[47] | BAC (>26,000) | <1000 | 161 NSCLC (103AC, 58 SCC) (correlated with mRNA microarray profiles) [38] |
[48] | BAC (4362) | 1000 | 20 NSCLC (11AC, 9 SCC) |
[49] | Multiple | >3400 NSCLC tumors and cell lines (from public repositories for 20 studies) | |
[50] | Oligo (244,000) | 2.1 | 123 Paired normal and NSCLC (57AC, 50 SCC, 13 LCC, 3 unclassified) |
Adenocarcinoma | |||
[51] | BAC (800) | 55AC | |
[52] | BAC (1440) | 2300 | 15AC |
[53] | SNP (500,000) | 575AC | |
[54] | BAC (2621) | 1000 | 3AC cell lines |
[55] | BAC (>26,000) | 400 | 26AC (non-mucinous BrAC, invasive AC with BrAC features) |
[56] | BAC (1440) | 2300 | 21AC (11 early response, 10 non-relapse) |
[57] | Oligo (385,806) | 6 | 138AC |
Squamous cell carcinoma | |||
[58] | BAC (1440) | 2300 | 14 SCC |
[59] | BAC (6500) | 35 SCC (with and without COPD) | |
[60] | Oligo | 1000 | 22 SCC |
[61] | BAC (>26,000) | <1000 | 52 SCC (with and without arsenic exposure) |
[62] | Oligo (244,000) | 2.1 | 62 SCC |
SCLC/neuroendocrine | |||
[63] | SNP (1494) | 17 SCLC | |
[24] | SNP (1494) | 33 Lung cancer cell lines (19 NSCLC, 14 SCLC) | |
[64] | 5p BAC (491) | 100 | 15 SCLC cell lines |
[65] | 1p BAC (642) | 15 SCLC cell lines | |
[66] | cDNA (39,632) | 70 | 24 SCLC cell lines |
[27] | cDNA (~10,000) | 20 NSCLC cell lines (11AC, 7 SCC, 2 LCC), 9 SCLC cell lines | |
[67] | BAC (800) | 10 SCLC, 31 LCNEC | |
[31] | SNP (115,593) | 51 NSCLC (37AC, 10 SCC, 4 BrAC); 26 NSCLC cell lines (14AC, 4 SCC, 4 LCC, 2 NSCLC, 2 BrAC, 1 AdSq); 19 SCLC tissues; 5 SCLC cell lines | |
[68] | BAC (32,433) | 14 SCLC cell lines, 6 normal cell lines | |
[69] | SNP (114,000) | 23.6 | 23 SCLC cell lines |
[42] | BAC (>26,000) | <1000 | 1AC, 1 SCLC, 1 LCNEC (from same patient) |
[43] | BAC (>26,000) | <1000 | 112 LC cell lines (77 NSCLC, 32 SCLC) (with and without EGFR TKI treatment) |
[70] | BAC (2464) | 46 SCLC, 5 SCLC cell lines | |
[71] | Oligo (180,000 and 105,000) | 13 SCLC cell lines | |
Likewise, loss-of-heterozygosity (LOH) events as indicators of inactivation on one allele of TSGs were more finely mapped using these high-throughput technologies. Nonetheless, the earlier polymorphic DNA marker-based technologies of Southern Blot and repetitive sequence PCR (e.g., microsatellite) contributed greatly to the localization and identification of candidate TSGs by identifying allele loss in tumor cell lines, primary tumor cells and preneoplastic cells associated with invasive cancers.
With the advent of newer technologies such as digital PCR and massively parallel sequencing (commonly referred to as next generation sequencing or NGS), comes even more capacity to detect and identify copy number variation in lung cancer. Rapid technological advances mean that NGS can be applied to the study of formalin-fixed, paraffin-embedded (FFPE) samples with relatively small quantities of DNA starting template [22], using next-generation sequencing for high resolution multiplex analysis of copy number variation from nanogram quantities of DNA from formalin-fixed, paraffin-embedded specimens. Indeed, NGS has largely superseded older techniques for detecting tumor CNVs.
28.3 Oncogenes and Growth Stimulation
Proto-oncogenes encode proteins that are positive effectors of the transformed phenotype and may be considered positive growth regulators. The normal cellular counterparts of these proto-oncogenes are often components of normal growth signaling pathways. Activation of these proto-oncogenes usually occurs via aberration of a single allele, including by gene amplification, point mutation and constitutive overexpression, leading to a gain in function or dominant effect. These proto-oncogene products include various growth factors, receptor tyrosine kinases, non-receptor tyrosine kinases, membrane-associated G proteins, cytoplasmic serine/threonine kinases, and nuclear transcription factors.
28.3.1 ERBB Receptor Tyrosine Kinases
The epidermal growth factor receptor or ErbB family of tyrosine kinases consists of four members—ErbB1 (EGFR), ErbB2, ErbB3, and ErbB4 [72]. The family of tyrosine kinases is important for many developmental and physiological processes. ERRB receptors are activated in response to p eptide binding resulting in receptor hetero- or homo dimerization [73]. The peptide growth factor ligands that interact with the ERBB family of transmembrane receptor tyrosine kinases are called neuregulins, neu differentiation factors, or heregulins [74]. The neuregulin and ERBB (ERBB2, ERBB3, and ERBB4) families may be considered to be potential growth stimulatory loops involved in the development of lung cancer [75]. On binding neuregulin, ERBB receptors homodimerize or heterodimerize, subsequently inducing intrinsic kinase activities that initiate intracellular signal transduction cascades such as the MAP kinase pathway. Although ERBB2 (also commonly known as ERBB2/neu) by itself lacks ligand binding ability, it plays a major coordinating role by enhancing and stabilizing dimerization. Each directly liganded receptor appears to dimerize preferentially with ERBB2, and resulting ERBB2-containing heterodimers have very high signaling potency. Activation of ERBB2 plays an important role in the development of many human cancers. The three major mechanisms leading to ERBB activation in cancer are gene amplification, altered ligand expression, and mutations in the receptor kinase or extracellular domain. The ERBB2 gene maps to chromosome 17q21, and its amplification and overexpression has been implicated in the development of several human cancers. Although amplification appears uncommon in lung cancer, ERBB2 is highly expressed in over a third of NSCLCs, especially in the adenocarcinoma subtype [76–78]. Experiments with transfected ERBB2 suggested that ERBB2 overexpression on its own was insufficient, but did contribute, to tumor induction in immortalized human bronchial epithelial cells [79]. Additionally, an anti-ERBB2 monoclonal antibody has been developed which can inhibit the in vitro growth of NSCLC cell lines expressing ERBB2 [80]. Some but not all studies have suggested that ERBB2 overexpression correlates with shorter survival in lung cancer [81–83]. In any case, other observations including the enhanced metastatic potential resulting from transfection of the ERBB2 gene into a NSCLC cell line in a xenograft model [84] support the idea that ERBB2 overexpression may be an adverse clinical indicator in some lung cancer patients. Transfection and overexpression of the ERBB2 gene in a constitutively low ERBB2 expressing NSCLC cell line also led to the induction of a drug resistant phenotype [85]. In vitro assays further suggest that ERBB2 overexpression may be associated with intrinsic multidrug resistance to chemotherapy agents in NSCLCs [86]. ERRB2 mutations in themselves are fairly rare in NSCLC [87, 88], with mutations being in-frame insertions situated in exon 20 (reminiscent of EGFR mutations). It is well recognized that increased copy numbers of ERBB2 in breast cancer predict response to trastuzumab [89]. Interestingly it appears that this may also be important for a group of non-small-cell lung cancer patients. ERBB2 amplification is found in 11 % of NSCLC specimens [90]. Clinical studies demonstrated that the subgroup of NSCLC patients with ERBB2 amplification achieve clinical benefit when trastuzumab is added to chemotherapy regimens [91].
28.3.2 EGFR
Another ERBB member, ERBB1 (better known as epidermal growth factor receptor, EGFR ), has a role in regulating epithelial proliferation and differentiation. Epidermal growth factor (EGF) and transforming growth factor-α (TGFα) are prominent amongst the six characterized mammalian ligands that bind to the EGFR receptor. While no ligand has yet been identified for ERBB2, it is the most frequently used partner in heterodimerization with EGFR. The production of EGFR ligands, especially TGFα by lung cancer cells expressing cognate receptors, has led to suggestions that this system represents an important autocrine loop in lung cancer [75, 92,,93]. Ligand binding to EGFR results in homodimerization or heterodimerization which initiates autophosphorylation of tyrosine residues in the intracellular domain [94]. This initial phosphorylation event is thought to trigger a phosphorylation mediated signal transduction cascade through two major pathways—the Ras/Raf/MAP kinase and the PI3K-Akt pathway. Ras/Raf/MAPK activates MAPK3 and MAPK1 transcription factors resulting in increased cell proliferation, survival, and transformation, while PI3K-Akt inactivates BCL2 family proteins and caspase 9, thus promoting survival in the face of apoptotic stimuli including radiation and DNA damage. Both pathways are implicated in development of malignancy [95]. Transgenic mice lacking EGFR have been generated. These mice develop abnormal epithelia in several organs, including the lung, in which there was impaired branching, deficient alveolization and septation [96, 97].
Activation of EGFR in lung cancer cells generally occurs by overexpression, with mutations and gene amplification being mechanistically implicated. Activation appears to be more common in NSCLC than in SCLC and may be related to tumor stage and differentiation [92, 98]. In NSCLC, amplification of EGFR is a common event resulting in overexpression in 60 % of metastatic NSCLC and is associated with an adverse prognosis [99–101]. In addition, in NSCLC there is increased expression of EGFR ligands (EGF and TGFα), which establish an autocrine loop of hyperactivation [102]. EGFR mutations of the tyrosine kinase domain are of two main types—either small in-frame deletions or missense substitutions clustered within the ATP binding pocket encoded by exons 18–21 [103–105]. Mutations occur more often in individuals with adenocarcinoma histology, East Asian origin, female gender, and never smoker status. In addition, they appear to be inversely correlated with KRAS mutations [106]. Mutations associated with tyrosine kinase inhibitor (TKI) responsiveness occur in the ATP-binding pocket of the tyrosine kinase domain of EGFR and are referred to as activating mutations [107]. In the presence of an activating mutation there is decreased affinity of this binding site for ATP and increased affinity for TKIs [108]. Interestingly, transgenic mice models have demonstrated that induction of activating EGFR mutations results in the development of adenocarcinoma like lung cancer [109]. Activating mutations are categorized into three classes: (1) class I mutations consist of deletions (in exon 19), (2) class II mutations consist of single nucleotide substitutions causing an amino acid change in exon 21, and (3) class III mutations consist of in-frame deletions or insertions (in exon 20). A 15 bp deletion involving amino acid residues leucine 747 to glutamic acid 749 (ΔLRE) in exon 19 and a point mutation substituting arginine for leucine at codon 858 (L858R) in exon 21, account for 44 and 41 % respectively of all NSCLC EGFR mutations [107, 110]. Preliminary evidence suggests that patients with the exon 19 mutation (ΔLRE) respond better to TKIs than patients with the exon 21 point mutation, L858R [111–113]. Further, there are polymorphisms associated with increased EGFR protein production (shorter CA-SSR1 length and variant forms of SNPs-216 and -191). These were found to be rare in East Asians as compared to other ethnicities, but in tumors from East Asian patients, EGFR mutations were found to favor the shorter CA-SSR1 allele, and selective amplification of the shorter allele of CA-SSR1 occurred frequently in tumors harboring a mutation [114]. Interestingly, another specific EGFR mutation, T790M, has been linked to acquired TKI resistance, and also to familial NSCLC [115–117] and was found to display a growth advantage over wild-type EGFR in a human bronchial epithelial (HBEC) cell line [118]. This immortalized HBEC model system which can be easily manipulated is likely to gain increasing prominence in the functional testing of candidate molecules [119].
Both types of activation mutations are associated with increased autophosphorylation of Tyr1068 at the C-terminal indicating increased EGF-dependent receptor activation [104]. Consistent with the association found between tumor p-Akt expression and gefitinib response, is mechanistic evidence that signaling through the antiapoptotic STAT/Akt may be an important downstream pathway of activation for mutated EGFR [120]. Parallels may be drawn with the clinical effectiveness of another TKI, imatinib mesylate, in chronic myeloid leukemia and gastrointestinal stromal tumors expressing activating c-Kit mutations, in which constitutive STAT activation is frequent. The story in lung cancer may be more complex, since several EGFR signaling pathway genes have been found to be mutated in NSCLC. EGFR and KRAS mutations are detected in ~10 % and 15–20 % of NSCLCs, respectively. Somatic mutations are much less common in ERBB2 (~2 %; exons 19 and 20) and HER4 (~2 %, exons 20, 23), the lipid kinase PIK3CA (~4 %; exon 9), and the serine/threonine kinase BRAF (~2 %; exons 11 and 15) [121]. Other rare mutations remain to be discovered for example in FGFR4 [121]. Most of these alterations have been found to be gain-of-function mutations and are not usually associated with co-mutations in the other genes apart from PIK3CA mutations.
There is evidence that some cancers may become dependent upon certain growth pathways or upon specific components of the pathway, possibly to the extent of requiring their signaling to maintain the malignant phenotype. This phenomenon has been described as a state of oncogene addiction of cancer cells. Thus, lung cancers most dependent upon signaling through EGFR and its downstream cascade may be more susceptible to drugs targeted to this pathway. Clinical responses to the EGFR TKIs, gefitinib and erlotinib, have been shown to be largely independent of expression of EGFR, but were discovered to be strongly associated with the presence of mutations in the EGFR tyrosine kinase domain [103–105] and possibly gene amplification [122–124]. Second-generation agents, such as afatinib, are also effective GFR TKIs, and have distinguishing properties such as being irreversible inhibitors.
Some experimental molecular studies provide evidence that oncogenic kinases produce both pro-survival and pro-apoptotic signals that decay at different rates upon oncogene inactivation; pro-survival signals are rapidly attenuated, whereas pro-apoptotic signals are relatively longer-lived. This differential signal decay creates a temporal window during which pro-apoptotic outputs from the oncogenic kinase predominate to actively promote tumor cell death upon kinase inhibition, known as oncogenic shock [101, 125].
28.3.3 EML4/ALK Fusion Genes
Chromosomal rearrangements involving the tyrosine kinase anaplastic lymphoma kinase (ALK) occur in cancers including NSCLC. EML4-ALK fusion was first discovered by screening a cDNA library derived from the tumor of a Japanese male patient with adenocarcinoma of the lung [126]. The EML4-ALK fusion represents an inversion on the short arm of chromosome 2 (Inv(2)(p21p23)) that joins exons 1–13 of EML4 (echinoderm microtubule associated protein-like 4) to exons 20–29 of ALK [126], resulting in a chimeric protein consisting of an EML4 N-terminus and an ALK C-terminus.
Since then, multiple other variants of EML-ALK have been reported, all of which encode the same cytoplasmic portion of ALK but contain different truncations of EML4 as well as rarer fusions of ALK with other partners such as TFG and KIF5B [127]. The aberrant activation of ALK signaling is consistent with the notion of oncogene addiction as it is associated with marked sensitivity to ALK inhibitors such as crizotinib [128, 129]. Understanding the role ALK fusion genes play in lung carcinogenesis has meant a relatively rapid translation of ALK-fusion targeted treatment to the clinic for the genetically definable subset of affected patients representing a few percent of NSCLC tumors and more common in female never smokers. Like EGFR mutations, these aberrations are the so-called actionable mutations which enable the concept of precision medicine to be realized.
28.3.4 ROS1 Fusion Genes
Another gene that has recently been discovered to be rearranged in a subset of NSCLC is ROS1, an orphan receptor tyrosine kinase which is phylogenetically related to ALK [130]. Like tumors with ALK1 fusion gene drivers, tumors with ROS1 fusions also appear more common in never smokers and are also sensitive to crizotinib [131].
28.3.5 RAS Signal Transduction Pathway
The RAS gene family represents oncogenes which are important in a subset of lung cancers. RAS genes encode 21 kDa proteins, members of a large family of proteins including rho, rac, rab that regulate cytoskeletal changes, vesicular and nuclear transport, and proliferation. Mutations in the RAS genes are common in several cancers, including lung cancer [132]. The RAS proto-oncogene family (KRAS, HRAS, and NRAS) is usually activated by point mutations at codons 12, 13, or 61. Mutations affect 15–20 % of all NSCLC and approximately 20–30 % of lung adenocarcinomas but are very uncommon in SCLC [133]. Mutations in KRAS account for approximately 90 % of the RAS mutations in lung adenocarcinomas, and 85 % of KRAS changes involve codon 12. In unstimulated cells, RAS is inactive. Following ligand binding, receptor tyrosine kinases signal the RAS protein by interaction with downstream molecules such as GRB2 and the guanine nucleotide exchange factor, SOS. Wild-type CDKN1A-RAS protein is able to bind to guanosine triphosphate (GTP) but also has intrinsic GTPase activity which can hydrolyze bound GTP to guanosine diphosphate (GDP). Active GTP-bound RAS stimulates a downstream cascade ending with MAP kinase which migrates to the cell nucleus and subsequently activates various transcription factors. When GTP is hydrolyzed to GDP, the molecule assumes its inactive configuration and the signal transduction pathway returns to its inactive state. When a RAS gene undergoes oncogenic missense mutations, the mutant CDKN1A-RAS oncoprotein loses its capability to hydrolyze GTP and the molecule can no longer switch back to its inactive configuration. The resultant inappropriate growth signal to the cell nucleus is thought to contribute to unrestrained cellular proliferation; effectively causing a gain of function. This model underscores the general concept that proto-oncogenes encode proteins with important regulatory functions and that oncogenic activation results in mutant proteins with altered function.
The majority (70 %) of KRAS codon 12 mutations are G-T transversions, with either cysteine (TGT) or valine (GTT) replacing the wild-type glycine (GGT). This type of mutation also affects the TP53 gene in lung cancer cells, and represents the type of DNA damage expected from bulky DNA adducts caused by the polycyclic hydrocarbons and nitrosamines in tobacco smoke [134]. The correlation of KRAS mutations with a smoking history further implicates a causative role of tobacco smoke carcinogens in the acquisition of these mutations [135]. KRAS mutations are associated with distinct clinical characteristics in patients with NSCLC. They occur more frequently in lung adenocarcinomas from former or current smokers [136]. Furthermore in early stage lung cancer, the presence of RAS mutations is predictive of early relapse [137]. The relationship of KRAS mutations to a poor prognosis in both early and late stage NSCLC is debated and may be technique related [138–146]. Although it has also been suggested that RAS mutations induce resistance to chemotherapy and radiation, no association between RAS mutation and in vitro resistance against a range of chemotherapeutic agents was found in a panel of NSCLC cell lines [147]. In lung adenocarcinomas the presence of KRAS mutations predict resistance to anti-EGFR therapies (gefitinib and erlotinib) [148]. Consequently in many centers routine testing for KRAS mutations in lung adenocarcinomas is performed to predict response to anti-EGFR therapy [149]. The reason for the predilection of RAS mutations for the adenocarcinoma histological subtype of lung cancers is unclear. One possible explanation is that the activation of certain oncogenes may result in disparate tumor differentiation pathways. To study this, various oncogenes have been introduced into a non-tumorigenic cell line BEAS-2B, derived from normal bronchial epithelial cells transfected with SV40. Overexpression of c-myc and c-raf-1 for instance resulted in the development of large tumor cells with certain neuroendocrine markers [150]. Indeed, the subtype specificity may be even more intriguing as a study has reported that KRAS mutations were seen in parenchymal, but not in bronchial adenocarcinomas, indicating genetic heterogeneity [151]. Furthermore, the goblet-cell subtype appears to have the highest frequency of KRAS mutations compared with other adenocarcinoma subtypes [152]. Some investigators have also suggested that rare alleles of the HRAS minisatellite locus represent a major risk factor for common types of cancer, including lung cancers, but this possibility has not been firmly established [153]. Considerable attention has been directed towards the development of RAS inhibitors, however to date none has achieved clinical utility [149]. Consequently, novel approaches to block posttranslational activation of RAS have been pursued, such as farnesyl transferase inhibitors and other inhibitors of downstream RAS effectors [149].
Another proto-oncogene, BRAF, which encodes a direct downstream effector of RAS may also be relevant to lung carcinogenesis. BRAF somatic missense mutations are common in malignant melanomas and are occasionally found in a range of human cancers. In lung cancer perhaps 3 % of NSCLC may have mutated BRAF1 [154–156]. Most melanoma BRAF mutations involve codon 599, but NSCLC BRAF mutations are different, most being non-V599. This may result in therapeutic differences between lung cancer and melanoma in response to RAF inhibitors, which are now subject to clinical trial.
The other signal transduction molecules downstream of RAF in this pathway include MEK (MAP kinase/ERK-activating kinase), and ERK (extracellular signal-regulated kinase) as well as their regulatory phosphatases (such as PP2A). Although constitutively active mutants of MEK were shown to be capable of transforming cells suggesting that MEK can function as a dominant oncogene, studies have shown that the MAP2K1 and MAP2K2 genes are only rarely mutated in lung cancer [157]. On the other hand, the mitogen-activated protein kinase kinase 4 (MKK4) gene, located approximately 10 cM centromeric of TP53 on 17p, has been found to be homozygously deleted in a NSCLC cell line, leading to speculation that it may be a candidate tumor suppressor [158].
28.3.6 FGFR1
Overall, unlike adenocarcinomas which have a number of actionable mutations (EGFR mutations, ALK and ROS1 fusions), these potential targets appear less common in the other major NSCLC subtype, SCC. Recently, focal fibroblast growth factor receptor 1 (FGFR1) amplification was discovered in squamous cell lung cancer (n = 155), and appears to be a relatively frequent finding (up to 22 % in an independent cohort) [159].
28.3.7 MYC
Stimulation of the RAS signal transduction cascade ultimately activates nuclear proto-oncogene products, including MYC which belongs to the basic helix–loop–helix leucine zipper (bHLH-LZ) class of transcription factors. MYC has been implicated in normal cell growth and proliferation through interaction with genes involved in DNA synthesis, RNA metabolism, and cell-cycle progression [160]. MYC proto-oncogenes are the cellular homologs of a gene present in several highly oncogenic avian retroviruses. Of the well-characterized myc genes, MYC is the most frequently activated in SCLC and NSCLC. On the other hand, its closely related cellular homologues, MYCN and MYCL1, are usually only activated in the SCLC subtype. In fact, MYCL1 was initially isolated from the DNA of a SCLC [161]. Activation of the myc genes has been observed by gene amplification or transcriptional dysregulation, leading to protein overexpression [162]. These genes may be amplified to 20–115 copies per cell, and in most cases, only one member of the MYC family is amplified. Gene amplification is generally associated with enhanced mRNA expression and increased protein production. A review of 17 different studies calculated that 36 of 200 (18 %) SCLC tumors and 38 of 122 (31 %) SCLC cell lines had gene amplification of one member of the MYC family [133]. In comparison, 25 of 320 (8 %) NSCLC tumors and 3 of 15 (20 %) NSCLC cell lines had MYC amplification. Thus, MYC family activation in general appears to be more frequent in SCLC than NSCLC. MYC amplification appeared to occur more frequently in cell lines which are often derived from metastatic lesions than in primary tumors, in patients previously treated with chemotherapy, and in the variant subtype SCLC [163]. These observations may help explain why MYC amplification has been reported to correlate with adverse survival. Rather than the translocation and point mutations seen in lymphomas, there have been reports of MYCL1 amplification with rearrangement in which MYCL1 fuses to the RLF gene, thereby resulting in a chimeric protein [164, 165]. In some cases, MYCL1 expression may be associated with neuroendocrine differentiation. For instance, all-trans-retinoic acid mediated growth inhibition in a SCLC cell line was associated with increased neuroendocrine differentiation and MYCL1 expression but decreased MYC expression [166].
28.3.8 Other Nuclear Proto-oncogenes
MYB, JUN, and FOS have also been implicated in lung cancer although their precise functional importance and biological role is still being investigated. JUN and FOS are heterodimeric proteins that function as immediate early transcription factors regulating cellular proliferation. There is conflicting data in lung cancer regarding their role in lung carcinogenesis. Some investigators have described higher expression of these genes in normal lung tissue adjacent to tumor than in the tumor itself. Conversely, other studies have reported higher expression in tumors with lack of expression in normal epithelium [167–170].
28.3.9 Oncogenes and Growth Stimulatory Loops
The hepatocyte growth factor/scatter factor (HGF/SF ) stimulates epithelial cells to proliferate, move and also carry out complex differentiation programs, such as morphogenesis and angiogenesis. HGF appears be a potent mitogen for normal and neoplastic bronchial epithelium [171]. During lung development, HGF levels increase during postnatal lung maturation and its receptors are expressed on bronchial and alveolar type II cells. HGF is involved in embryonal lung budding and branching, and stimulates mitogenesis and/or motogenesis of human bronchial epithelial, and alveolar type II, and SCLC cells in vitro. HGF is expressed at very low levels in normal lung but these levels increase in response to local lung or distant injury [172]. The MET proto-oncogene which encodes the HGF receptor was generally expressed in normal lung, as well as in SCLC and NSCLC. On the other hand, HGF was expressed in many NSCLCs but not in SCLCs, thereby indicative of an autocrine loop in the former [173–175]. A study of resected lung cancer tissue showed MET expression in 34 of 47 adenocarcinomas and 20 of 52 squamous carcinomas by western blotting and immunohistochemistry [176]. This study also suggested a poorer prognosis for tumors expressing the receptor especially for adenocarcinomas. Western blotting of proteins extracted from 56 NSCLCs (predominantly adenocarcinomas) using a polyclonal anti-HGF showed that high levels of immunoreactive HGF were associated with poorer survival for stage I tumors [177]. Recently, a gefitinib-sensitive lung cancer cell line that developed resistance to gefitinib as a result of focal amplification of the MET proto-oncogene was reported, also MET amplification was detected in 4 of 18 (22 %) lung cancer specimens that had developed resistance to gefitinib or erlotinib [178]. This suggests that MET amplification causes gefitinib resistance by driving ERBB3 (HER3)–dependent activation of PI3K, a pathway thought to be specific to EGFR/ERBB family receptors.
Another autocrine growth loop may involve the insulin-like growth factors , IGF-I and IGF-2, and the type I IGF receptor, IGF-R, which are frequently co-expressed in both SCLC and NSCLC [179]. Of the insulin-like growth factor family, it appears that IGF-2 may be the predominant member involved with the autocrine growth stimulation of lung cancer. The KIT proto-oncogene which encodes a tyrosine kinase receptor and its ligand, stem cell factor (SCF), are co-expressed in many SCLCs, and may thus represent another autocrine loop for lung cancers [180, 181]. In SCLC, activation of this putative SCF/KIT autocrine loop could conceivably provide a growth advantage or mediate chemoattraction. Unlike gastrointestinal stromal tumors, mutations in KIT are rare, although KIT expression can be demonstrated in SCLCs [182–184]. Platelet-derived growth factor (PDGFB), which is the proto-oncogene counterpart of v-sis, and its receptor (PDGFBR) were also found to be co-expressed in lung cancer, generating another potential autocrine loop [185].
In essence, many proto-oncogenes encode growth factors, regulatory peptides or their receptors and are expressed by lung cancer cells or adjacent normal cells, thus providing a number of autocrine or paracrine growth stimulatory loops [186]. Other autocrine systems not obviously involving established proto-oncogenes also exist. In fact, the autocrine loop comprising gastrin-releasing peptide, other bombesin-like peptides (GRP/BN) and their receptors is arguably the best characterized growth stimulatory loop in lung cancer. GRP/BN has been associated with many physiologic effects including regulation of secretion, growth, and neuromodulation. There are three human GRP/BN receptor subtypes which belong to the G-protein coupled receptor superfamily with seven predicted transmembrane domains [187]. The cellular responses of SCLC to GRP/BN stimulation have been extensively studied [188]. Preliminary data also suggests that GRP/BN may regulate the MAP kinase Cascade, at least in certain tumor cells [189]. IHC studies showed that approximately 20–60 % of SCLC cancers expressed GRP, while NSCLCs expressed GRP less frequently [133]. In comparison, expression of the three GRP/BN receptors is widespread in both SCLC and NSCLC cell lines, with most cell lines expressing at least one of the three receptors and many cell lines expressing more than one receptor [187].
28.3.10 BCL2 Proto-oncogene and Apoptosis
Tumor cells can acquire the ability to escape apoptotic pathway by which normal cells would usually undergo programmed cell death (apoptosis) in response to appropriate conditions such as DNA damage. A large and growing nu mber of apoptosis regulatory gene products are classifiable into cell death agonists (Bax, Bak, Bcl-XS, Bad, Bid, Bik, Hrk) or antagonists (BCL2 , Bcl-XL, Bcl-w, Bfl-1, Brag-1, Mcl-1 and A1) [190]. Two key members of the apoptotic pathway are the BCL2 proto-oncogene product and the TP53 tumor suppressor gene product. BCL2 antagonizes the induction of programmed cell death by TP53. By protecting cells from the apoptotic process, BCL2 probably plays a role in determining the chemotherapy response through repression of apoptosis in cancer cells. A BCL2-transfected human SCLC cell line showed higher resistance to some anticancer agents by inhibiting apoptosis [191], and SCLC cells transfected with antisense oligodeoxynucleotides to BCL2 mRNA showed reduced cell viability with decreased BCL2 levels facilitating apoptosis [192]. BCL2 expression may correlate with neuroendocrine differentiation [193]. BCL2 expression is also relatively higher in squamous cell carcinoma (25–35 %) than in adenocarcinoma (~10 %) [193–196]. An inverse relationship between immunohistochemical BCL2 expression and abnormal TP53 expression in resected NSCLCs has been reported [197, 198]. These results suggest the hypothesis that either TP53 mutation or upregulation of BCL2 expression is sufficient to modify the apoptotic pathway in NSCLC, and that BCL2 positive tumors may show less aggressive behavior [197]. In contrast, SCLCs usually have both TP53 mutations and BCL2 overexpression. Previous IHC studies of BCL2 in lung cancer have shown highest expression in small cell cancers. For instance BCL2 protein is immunohistochemically expressed in most SCLCs (75–95 %) [193, 199, 200]. This observation initially seems inconsistent with the finding that SCLCs are often much more sensitive than NSCLCs to chemotherapy; a situation in which tumor death usually occurs by apoptosis. In addition, there was a paradoxical trend, albeit non-statistically significant, towards longer survival in patients whose SCLC tumors express BCL2 [200]. Likewise, a better survival of BCL2 positive lung cancer cases was observed in NSCLC [194–197]. Thus the role of BCL2 in lung cancer is complex and likely to further unfold. Notably, the suggestion that BCL2 may be converted to BAX-like death effectors by the caspase family of cysteine proteases may be relevant [201]. Alternatively, increased BCL2 immunoreactivity may possibly indicate reduced rather than enhanced function, similar to the situation of overexpression of nonfunctional mutated TP53. A BCL2 related protein called BAX promotes apoptosis and may act as a tumor suppressor [202]. Furthermore, BAX may be a downstream transcription target of the TP53 pathway. BAX complexes with BCL2 to form homodimers or heterodimers, and it has been suggested that the BCL2–BAX ratio determines cellular apoptotic susceptibility. For instance, the immunohistochemical staining of BAX and BCL2 were inversely related in 121 neuroendocrine (NE) lung cancers. Most carcinoids showed low BCL2 and high BAX expression in contrast to the inverse situation in most SCLCs and large cell NE cancers [203]. This would potentially lead to a higher degree of apoptosis in carcinoids compared to SCLCs, correlating with clinical behavior where carcinoids are markedly less metastatic than SCLC.
28.4 Tumor Suppressor Genes and Growth Inhibition
28.4.1 Genetic Loss in Lung Cancer
Apart from aneuploidy, lung cancer c ells are also characterized by many other structural cytogenetic abnormalities including deletions and nonreciprocal translocations. Frequent deletions of chromosome region 3p14-23 was one of the initial cytogenetic observations made in SCLC, this finding was later found to be also applicable to NSCLC [204]. Subsequently, many specific deletions at a range of chromosomal regions have been revealed by cytogenetics, molecular allelotyping, and chromosomal and array CGH, data which suggests the presence of underlying tumor suppressor genes in lung cancers.
TSG (e.g., TP53, RB1) products are negative growth regulators. It is their loss of function, classically by loss of one allele and point mutation of the other allele, which contributes to malignant transformation. The requirement for the mutations to affect both alleles of the tumor suppressor gene represents the two-hit hypothesis initially proposed by Knudson for retinoblastomas. Using allelotyping, chromosomal regions 1p, 1q, 3p (several sites), 5q (APC/MCC cluster), 8p, 9p21 (CDKN2A), 11p13, 11p15, 13q14 (RB1), 17p13 (TP53), and 22q as well as several other sites have been found to be frequently involved in lung cancer cells or cell lines derived from lung cancer tissues [205]. This leads to the notion that if most of these sites encode tumor suppressor genes, then individual tumors must have acquired inactivation of multiple genes to become clinically evident. Some of these are common to both SCLC and NSCLC, and some are more frequent in a given histologic type. The best characterized of these deletions appear to target genes which are now accepted as classical tumor suppressor genes such as TP53, RB1, and CDKN2A. On the other hand, there is also mounting evidence that there may be other closely situated genes that are also affected by the chromosomal deletions, particularly as some of these deletions may be quite large and even involve the whole chromosomal arm or chromosome.
28.4.2 TP53
Mutations in the TP53 gene are the most common genetic alteration found in human cancers. The TP53 gene encodes a protein that functions as a transcription factor, particularly in response to DNA damage by γ or ultraviolet irradiation and carcinogens [206]. It has been called the guardian of the genome and the guardian of the G1 checkpoint. TP53 is believed to play a major role in maintaining the integrity of the genome since loss of TP53 function allows inappropriate survival of genetically damaged cells, leading to the evolution of a cancer cell. DNA damage is a major upstream event in TP53 activation and results in a rapid increase in the level of TP53 protein, and activation of TP53 as a sequence specific transcription factor regulating expression of downstream genes. The net effect is either a stop to cell cycle progression to permit repair, or apoptosis if the damage is too great. TP53 can itself detect and bind sites of primary DNA damage, using its C-terminal domain. Hypoxia is also able to stimulate TP53 levels and lead to apoptotic cell death [207]. Oxygen delivery and blood supply becomes rate-limiting when a tumor reaches a critical size and tumor. Hypoxia may thus act as a physiological selective agent against apoptosis-competent cells in tumors. On the other hand, such selection may allow for the expansion of clones with acquired defects in their apoptotic program genes. The genes downstream of TP53 include CDKN1A (also called WAF1/CIP1), MDM2, GADD45A (growth arrest and DNA damage-inducible), BAX, IGF-BP2, and cyclin G, which participate in controlling cell cycle arrest at the G1/S phase transition and apoptosis. A link between mutant TP53 and aneuploidy has been revealed by studies implicating TP53 as an active component of a mitotic spindle checkpoint and as a regulator of centrosome function . Thus, TP53 appears to participate in the DNA damage checkpoints of the cell cycle at both the G1/S transition and at the G2/M boundary.
The TP53 gene plays a critical role in lung cancer as well as in many other types of cancers, and genomic interrogation indicates consistent features across tumor types [208]. Many somatic TP53 mutations in human tumors and cell lines have been published, and compiled into large databases. In both SCLC and NSCLC, one copy of the chromosomal region 17p13 which contains TP53 is frequently deleted; one hit. Structural abnormalities in TP53 and also p16 occur commonly in lung cancer, as shown by structural aberrations on FISH analysis (especially breaks and loss) [209]. Mutational inactivation of the remaining allele, the second hit, occurs in 75–100 % of SCLCs and ~50 % of NSCLCs [134], leading to loss of TP53 function. Although found throughout the entire coding region, TP53 mutations in lung cancer are most common in the evolutionarily conserved exons 5–8. The types of TP53 mutations include missense and nonsense mutations, splicing abnormalities, as well as larger deletions. Evidence supporting a causative role for tobacco smoke in inducing TP53 mutations comes from observing that TP53 mutations in lung tumors correlate with cigarette smoking, and that the most common TP53 mutations in lung cancer are the G-T transversions expected from tobacco smoke carcinogens [134]. Additional evidence for a pulmonary oncogenic role for TP53 dysfunction comes from the finding that transgenic mutant TP53 mice develop lung cancers in addition to bone and lymphoid tumors [210]. Furthermore, reintroducing a wild-type TP53 gene into lung cancer cells dramatically blocked tumor cell growth due to apoptosis (not G1 arrest) despite concurrent abnormalities of several other tumor suppressor genes and oncogenes [211, 212].
Many TP53 mutations are missense mutations which prolong the half-life of the TP53 protein to several hours, leading to increased protein levels which can be detected by immunohistochemistry as a surrogate for molecular analysis [213]. IHC studies have shown abnormal TP53 expression in 40–70 % of SCLCs and 40–60 % of NSCLCs [203, 214–217]. Most studies have shown that the frequency of TP53 overexpression is higher in squamous cell carcinomas than in adenocarcinomas. The predictive value of TP53 mutations for survival, whether assayed by immunohistochemistry or by molecular analysis, is controversial. A summary of 14 studies of the prognostic importance of TP53 mutations or overexpression in NSCLC (mutational analysis (4 studies); immunostaining (8 studies) and both techniques (2 studies) yielded controversial results [218]. TP53 mutations predicted shortened survival in half of the four reported mutational analyses, whereas the other two found no such difference. Of the 10 IHC studies, aberrant TP53 expression was associated with a shortened survival in five studies, an improved survival in three studies, and no survival effect in two studies. In one study that simultaneously analyzed both mutations and protein expression , TP53 overexpression but not gene mutation predicted shortened survival. Perhaps the various TP53 mutants or types of wild-type TP53 overexpression have different effects on lung cancer behavior. Alternatively, wild-type TP53 expression may be immunohistochemically detectable in certain tumors. Finally, different antibodies which may not be strictly comparable for detecting aberrant TP53 expression have often been used.
In certain cancers, such as that of the uterine cervix, TP53 can be alternatively inactivated through binding of the oncogenic E6 protein of human papilloma virus (HPV) to the TP53 protein; a process which inactivates its tumor suppressor activity by promoting TP53 degradation. The epitheliotropic HPV may also be involved in some respiratory tract lesions, for example, HPV subtypes 6 and 11 have been associated with most cases of tracheal and bronchial papillomatosis. While there are reports of neoplastic transformation of these benign papilloma, it has also been suggested that HPV may also play a part in the development of de novo bronchogenic carcinomas [219–221]. Morphological studies have shown occasional presence of HPV-suggestive lesions in primary squamous cell carcinomas, and DNA hybridization studies to detect HPV DNA in lung cancers show conflicting results, ranging from 0 to 40 % [222–224]. Most PCR studies looking for HPV sequences suggest that any potential involvement of HPV in primary lung cancer is likely to be limited [225–228] although some investigators have found more frequent involvement using in-situ hybridization and PCR [229].
CDKN1A , also known as WAF1 (wild-type TP53-activated fragment 1), CIP1, or p21, is a TP53-responsive gene, which inhibits cyclin/cyclin dependent kinase complexes in the G1 phase of the cell cycle as well as proliferating cell nuclear antigen (PCNA). Although not somatically mutated in lung cancer [230], CDKN1A RNA and protein overexpression was seen in ~65 % of NSCLC cases, especially in well differentiated tumors. This high frequency suggests that CDKN1A can be expressed independently of TP53 gene/protein alterations which are so fr equent in lung cancers [231]. A case control study has suggested that a C to A codon 31 polymorphism (ser- > arg) in CDKN1A is associated with the development of lung cancer [232].
MDM2 is an oncoprotein that can inhibit both TP53 and RB1. By binding its transcriptional activation domain, MDM blocks the ability of TP53 to regulate target genes. It also causes rapid reduction of TP53 levels through enhanced proteasome-dependent degradation. Conversely, TP53 activates the expression of the MDM2 gene in an autoregulatory feedback loop. However, phosphorylation of TP53 by DNA-dependent protein kinase (DNA-PK) after DNA damage leads to reduced interaction of TP53 with MDM2, most likely due to a TP53 conformational change [233]. In some human sarcomas and brain tumors, the chromosome 12q MDM2 gene is amplified and its protein overexpressed. In lung cancer MDM2 gene amplification was only detected in 2 of 30 NSCLCs. Nonetheless, these investigators also found MDM2 protein expression in 48 of 201 NSCLCs by immunohistochemistry and suggested that MDM2 expression without abnormal TP53 expression was a favorable prognostic factor [234]. Sp1 can further activate MDM2 and repress TP53, and this is associated with overexpression of DNA 5′-cytosine-methyltransferase1 (DNMT1), which can epigenetically dysregulate tumor suppressor genes [235].
A gene encoding TP73 , a protein that shares considerable homology with TP53 was mapped to 1p36 [236]. This is of interest as 1p36 is also a site of frequent allelic deletion in lung and other cancer cells [205]. Although TP73 mutations were not detected in neuroblastomas despite frequent LOH, TP73 can activate the transcription of TP53-responsive genes and inhibit cell growth in a TP53-like manner by inducing apoptosis [237].
28.4.3 The RB1/Cyclin D1/CDK4/CDKN2A Pathway
28.4.3.1 The Retinoblastoma (RB1) Gene
The retinoblastoma (RB1 ) gene located in chromosomal region 13q14 encodes a nuclear phosphoprotein that was initially identified as a tumor suppressor gene in retinoblastomas. The RB1/cyclin D1/CDK4/CDKN2A pathway is central to the regulation of the G1 to S phase transition of the cell cycle. Hypophosphorylated RB1 binds and controls other cellular proteins including the transcription factor E2F which is essential for the G1/S phase transition when bound to E2F sites in cooperation with the DP family of transcription factors. Transcriptional activation is mediated by free E2F whereas hypophosphorylated RB1 antagonizes heterodimers formed by E2F and DP, thereby resulting in inhibition of S phase entry. Cyclin D1/cyclin dependent kinase 4 (CDK4) and other cyclin/CDK complexes phosphorylate RB1 with subsequent loss of its binding pocket activity that is needed to sequester the transcription factors, thereby releasing E2F and allowing entry into S phase. It is becoming more apparent that one of the four genes responsible for RB1/cyclin D1/CDK4/CDKN2A pathway is mutated or functionally altered in many cancers including lung cancers. It has also been shown that the RAS signaling pathway may functionally link to cell cycle regulation by RB1 [238]. Furthermore, RB1 also appears to have other functions; it can for instance repress transcription of all three nuclear RNA polymerases classes (Pol I, Pol II, and Pol III) [239]. RB1 also appears to inhibit apoptosis and may be actively involved in induction of differentiation. In terms of interacting partners for RB1, overexpression of E2F1 or E2F1/TFDP1 cooperates with activated ras in fibroblast transformation assays, and these transformed cells can form tumors in nude mice. Studies of E2F1 deficient transgenic mice have however suggested that E2F1 may also have a tumor-suppression function since mice lacking E2F1 developed a broad spectrum of tumors including highly invasive lung adenocarcinomas [240]. Nonetheless, studies of E2F1 in lung cancer cells have not yet been reported.
RB1 mutations together with loss of the wild-type allele have been consistently demonstrated in lung cancers [241, 242]. The RB1 protein is abnormal in over 90 % of SCLCs and 15–30 % of NSCLCs [243–245]. RB1 mutations in lung cancers include truncation by deletions, nonsense mutations, or splicing abnormalities. There are only a few studies of RB1 point mutations in lung cancer at least partly due to its 200 kb genomic size and 27 exon structure. However, from these limited studies, most mutations result in RB1 truncation [246], although a rare missense mutation in the RB1 pocket domain has been shown to cause defective RB1 phosphorylation and binding to oncoproteins [247]. The sensitivity for detecting RB1 abnormalities in SCLCs varies by detection method: ~20 % by Southern blot analysis detecting band loss; ~60 % by Northern blot analysis detecting absent or abnormal RNA; and ~90 % by protein or immunohistochemical (IHC) analysis. Thus RB1 abnormalities are very frequent in SCLC, particularly in comparison to NSCLC. In NSCLC, a large study showed RB1 abnormalities in 2/219 (0.9 %) by Southern analysis, 22/219 (10 %) by Northern analysis, and 53/163 (32 %) by IHC [243]. The absence of RB1 expression was associated with poor prognosis in NSCLCs, particularly stage I and II disease, in some but not all studies [245, 248–250]. On the other hand, the observation of frequent LOH on chromosome 13q but relatively less frequent RB1 inactivation in NSCLCs, have prompted the notion that the allele loss on 13q targets other tumor suppressor genes on this chromosome apart from RB1 [251].
The relatives of retinoblastoma patients who are also germ line carriers of an RB1 mutation, are about 15 times more likely to die from lung cancer than the general population [252]. Furthermore, it has been shown that reintroduction of a wild-type RB1 gene led to growth suppression of SCLC cells [253]. There are two other members of the RB1 gene family, p107 and pRB2/p130, which are structurally and functionally related genes. They have also been implicated in lung cancer to a limited extent [254]. One SCLC cell line was shown to have a point mutation of p130 in a splice acceptor site leading to loss of exon 2 and production of a truncated p130 protein [255].
28.4.3.2 Cyclin D1 and CDK4
The relatively infrequent involvement of RB1 in NSCLC compared to SCLC suggested that alternative members of this growth suppressive pathway might be affected. As cyclin D1 inhibits the activity of RB1 by stimul ating its phosphorylation by CDK4, cyclin D1 overexpression was an attractive candidate for disrupting the RB1 growth control pathway. In keeping with this, cyclin D1 was found to be overexpressed in some NSCLCs with normal RB1 protein expression [256, 257]. Cyclin D1 is encoded by the CCND1 (also known as PRAD1 or BCL1) proto-oncogene which is situated on chromosome 11q13. Compared with an immortalized bronchoepithelial cell line, cyclin D1 was overexpressed by several to 100-fold in 11/12 NSCLC cell lines [256]. Amplification of cyclin D1 was detected in 15 % and overexpression in 47 % of 53 primary NSCLC tumors [257]. Cyclin D1 immunohistochemical overexpression has been reported to correlate with Ki67 labeling and with patient survival [258, 259]. Abnormal immunostaining of CCND1 and RB1 has also been frequently seen in epithelial cells from the resection margin of lung cancers, raising speculation that these changes may be relatively early events in lung carcinogenesis [260]. Although gene amplification of CDK4 has been reported in 10–15 % of malignant gliomas and certain other malignancies, its role in lung cancer has not yet been reported. Interestingly, the chromosome 12q13-15 region which harbors the CDK4 gene also contains the MDM2 locus. While CDK4 and MDM2 often show co-amplification in sarcomas and glioblastomas, some tumors show only CDK4 amplification but not MDM2 amplification, and vice versa, indicating that each gene is an independent amplification target [261].
28.4.3.3 CDKN2A and Other CDK Inhibitor Genes
The CDKN2A protein functions as a cell cycle modulator which appears to regulate RB1 function by inhibiting CDK4:cyclin D1 kinase activity, and represents another important genetic target for disrupting the RB1/cyclin D1/CDK4/CDKN2A pathway. The CDKN2A (or MTS1) tumor suppressor gene is situated at chromosome 9CDKN1A. The short arm of chromosome 9, including 9CDKN1A frequently undergoes allele loss and mutation in a variety of human cancers including lung cancer [262–265]. A summary of a wide variety of cancers identified several mutational hot spots (point and other mutations including deletions, insertions and splice mutations), including some at conserved residues within the ankyrin domains of CDKN2A [266].
CDKN2A abnormalities have been extensively reported in lung cancer and are found frequently in NSCLC but rarely in SCLC. Homozygous deletion or point mutations have been observed in 10–40 % of NSCLCs [267–275]. Absent expression of CDKN2A was detected by Northern blot, Western blot or IHC analyses in 30–70 % of NSCLC. Epigenetic hypermethylation of 5′ CpG islands cause the functional downregulation of CDKN2A in lung cancers carrying no genetic mutations of CDKN2A [276, 277]. The multiple mechanisms of CDKN2A inactivation may account for the relatively low rates of inactivating deletions and point mutations seen in the earlier genetic studies [278]. Since higher frequencies of deletions or mutations have been observed in cultured cell lines and metastatic sites compared to primary lesions, it has been suggested that CDKN2A mutations may be associated with tumor progression and more advanced lung cancer [279]. While an IHC study of primary NSCLCs demonstrated an association of CDKN2A -negativity with more advanced with clinical stage [250], this finding was not confirmed in another study [280]. Nevertheless, both studies showed that about 30–40 % of early stage primary NSCLCs had absent CDKN2A expression. Finally, there is conflicting data as to whether the absence of CDKN2A expression is a predictor of adverse survival in NSCLC [250, 281]. CDKN2A abnormalities are perhaps the most common mechanism for inactivating the RB1/cyclin D1/CDK4/CDKN2A cell cycle control pathway in NSCLC. Conversely, direct RB1 inactivation appears to be the preferred mechanism in SCLC. Consequently, lung cancers are in general characterized by either RB1 inactivation (~90 % of SCLC and 15–30 % of NSCLC) or CDKN2A inactivation (30–70 % of NSCLC); either scenario leading to loss of this growth inhibitory pathway. The apparently mutually exclusive inactivation of either RB1 or CDKN2A has been well documented in NSCLC case series [250, 280, 282–286]. Furthermore, the simultaneous inactivation of both RB1 and CDKN2A is uncommon, but cyclin D1 overexpression can coexist with each of these abnormalities [278]. It is also noteworthy that a significant proportion of NSCLCs (10–30 %) appear to be normal for both RB1 and CDKN2A, thereby implicating cyclin D1 and CDK4 alterations.
The CDKN2A locus also encodes a second protein product which originates from an unrelated exon of CDKN2A (exon 1b) spliced onto exon 2 in an alternate reading frame (human p16b, murine p19ARF) [287]. Thus, exon 2 which is often deleted or mutated in NSCLC, is common to both CDKN2A and p19ARF. Intriguingly, mice lacking p19ARF but expressing functional CDKN2A were prone to tumor development, possibly through the TP53 pathway as TP53-negative cell lines were resistant to p19ARF-induced growth arrest [288]. Thus the extent of p19ARF damage through deletions and mutations to the CDKN2A locus, and its possible contribution to human lung carcinogenesis requires further investigation.
There are a number of other CDK inhibitor genes including CDKN2B, CDKN2C, CDKN2D, CDKN1A, CDKN1B, and CDKN1C. However, apart from CDKN2B, mutational analyses of these genes have not detected significant genetic changes in lung cancer. CDKN2B (also called CDKN2B or MTS2) shares ~70 % amino acid similarity and is situated immediately centromeric to CDKN2A. It also functions to restrain cell growth, probably by acting as an effector of TGFβ-mediated cell cycle arrest [289]. Co-deletion of CDKN2B and CDKN2A frequently occurs in NSCLC but point mutations targeting CDKN2B itself appear to be uncommon. The CDKN2C and CDKN2D genes have not been shown to be mutated in lung cancer [290]. Similarly, CDKN1B has not been found to be mutated in lung cancers [291], but low levels of CDKN1B protein were associated with a poor outcome in NSCLC patients [292]. The CDKN1C CDK inhibitor which maps to 11p15, is usually imprinted with expression of the maternal allele only. Thus, CDKN1C expression can be downregulated by selective loss of the maternal alleles, and this was found to occur in 11/13 lung cancer cases with LOH of 11p15 [293]. As point mutations have not been described in lung cancer, one could speculate that loss of a single allele may nonetheless represent the second hit needed to inactivate the imprinted CDKN1C gene.
28.4.3.4 Candidate 3p Tumor Suppressor Genes
It has long been known that chromosome 3p deletions oc cur commonly in cancers, notably lung and renal cancers. The very frequent deletion of one copy of the short arm of chromosome 3 in both SCLC (>90 %) and NSCLC (>80 %) has provided a strong basis for the hypothesis that one or more lung cancer tumor suppressor genes exist on this chromosomal arm. The karyotypes of most SCLC and many NSCLC have a del(3p) in addition to other often complex changes. These cytogenetic 3p deletions were subsequently confirmed by allelotyping which showed allelic loss not only in invasive cancers, but also in preneoplastic respiratory epithelial lesions associated with NSCLC [204, 294, 295]. The three distinct 3p regions have been identified by allelotyping are 3p25-26, 3CDKN1A.3-22, and 3p14-cen, consistent with the notion that there are probably three (or more) different tumor suppressor genes located on 3p [296]. In addition, as loss of both alleles resulting in homozygous deletions are thought to be strong markers for the locations of tumor suppressor genes, it is notable that five separate homozygously deleted regions have also been found in several lung cancer cell lines. There is one in the 3p14.2 region (FHIT gene location), one at 3p12-13 (U2020 cell line deletion) [297, 298], and three at the 3CDKN1A region.
The FHIT gene comprising 10 exons encoding a 1.1-kb transcript, maps to 3p14.2 and encompasses approximately 1 Mb of genomic DNA which includes the human common fragile site (FRA3B) and the t(3:8) translocation breakpoint of familial renal cell carcinoma. FRA3B is the most frequent of the common fragile sites which are chromosomal sites prone to breakages under stress, such as aphidicolin treatment. FHIT is a candidate tumor suppressor gene for lung cancers on the basis of frequent loss of heterozygosity in lung cancer and homozygous deletion in several lung cancer cell lines; the latter particularly affecting NSCLC [299–301]. Although reverse transcriptase PCR showed that 40–80 % of lung cancers demonstrated aberrant FHIT transcripts, these were nearly always also accompanied by wild-type FHIT transcripts [299–301]. As in other cancers, point mutations of FHIT appear to be rare in lung [299, 300], and there were initial concerns that the deletions noted merely represented the susceptibility of the FRA3B fragile site to breakages. Nevertheless, although FHIT abnormalities differ from the mutations and loss of wild-type transcript expression expected of classic tumor suppressor genes, absence of FHIT protein in primary lung tumors and cell lines correlating to DNA and/or RNA abnormalities has been detected by Western blot and immunocytochemical analyses [302]. Additionally reintroduction of exogenous wild-type FHIT suppressed tumorigenicity in nude mice of human cancer cell lines including a NSCLC cell line [303]. Furthermore, the much more frequent FHIT allele loss in lung cancers from smokers (80 %) compared to nonsmokers (22 %) suggests that this chromosomal region is selectively targeted by the carcinogens in tobacco smoke [304].
The 3CDKN1A.3 region has been extensively examined for putative tumor suppressor genes by several groups. The finding of several homozygously deleted lung cancer cell lines suggested a role for two distinct 3CDKN1A.3 regions. One region is defined by three distinct homozygous deleted SCLC cell lines with a minimum common deleted region mapped to span 370 kb [305–307]. The other 3CDKN1A region illustrated by several homozygous deletions has an estimated ~800 kb deletion [308]. Apart from these regions, hMLH1 (the human homolog of the yeast mutL gene) also resides on chromosome 3CDKN1A and while mutations of the hMLH1 gene have been found in hereditary colon cancer, lung cancer studies have not been reported [309]. Other well characterized candidate 3CDKN1A TSGs are SEMA3B and SEMAF, RASSF1A, and FUS1 [310]. SEMA3B and SEMAF are close to the other genes and part of the semaphorin axonal guidance gene family, both encode secreted proteins [311]. RASSF1A is frequently hypermethylated in lung cancer but rarely mutated. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network, is conserved in mammalian cells, and may be involved in controlling mitotic exit [312]. Fus1 is next to RSSF1A and while mutation of FUS1 is infrequent in lung cancers, protein under expression has been reported and exogenous overexpression of Fus1 protein inhibited colon formation in some NSCLC cell lines [313].
Other candidate 3p lung cancer suppressor genes include the von Hippel–Lindau (VHL) tumor suppressor gene at 3p25 which is frequently mutated in renal cell carcinoma but only uncommonly involved in lung cancers [314]. Abnormalities of retinoic acid receptors (RARs) have also been implicated in lung cancer pathogenesis. Several studies have indicated abnormalities of the expression or function of the RARB gene which maps to chromosome region 3p24, another site of frequent allele loss in lung cancer [315–320]. Mutational analysis however has failed to demonstrate mutations in RARB. Additionally, the TGFβ-type II receptor (TGFβ RII) gene at 3p22 is another candidate tumor suppressor gene as discussed below.
28.4.3.5 LKB1/STK11
Frequent losses of chromosome 19p in lung adenocarcinomas led to the fine mapping of the short arm of chromosome 19 and the discovery of the LKB1/STK11 gene which mapped in the minimal-deleted region [321]. Germ-line mutations at LKB1/STK11 result in the Peutz–Jeghers syndrome and an increased risk of cancer, and there is a relatively high frequency of somatic alterations (mainly nonsense mutations) in primary lung adenocarcinomas and in lung cancer cell lines [321]. These mutations may be linked to KRAS mutations and smoking and male gender in a subset of poorly differentiated lung adenocarcinomas, and are associated with transcriptional deregulation of molecules involved in signal transduction (e.g., FRAP1/mTOR, ARAF1, and ROCK2), cytoskeleton (e.g., MPP1), transcription factors (e.g., MEIS2, ATF5), metabolism of AMP (AMPD3 and APRT), and ubiquitination (e.g., USP16 and UBE2L3).
28.4.3.6 Other Candidate Tumor Suppressor Gene Locations
Apart from the known and candidate tumor suppressor gene locations discussed above, cytogenetic and allelotyping studies have shown allelic loss of many other chromosomal regions in lung cancer, thereby implicating involvement of other tumor suppressor genes. The chromosomal regions include 1p, 1q, 2q, 5q, 6p, 6q, 8p, 8q, 10q, 11p, 11q, 14q, 17q, 18q, and 22q. CGH and array CGH are helping to more precisely define these regions (Table 28.1).
Several of these chromosomal regions contain known or candidate tumor suppressor genes (such as WT1 at 11p13, DCC at 18q21, NF2 at 22q12), but these genes have not been found to be mutated in lung cancer [322, 323]. Regions on chromosome 5q, around the APC and MCC gene cluster are also frequently deleted in both subtypes of lung cancer. However, APC mutations have not been detected in lung cancer [323, 324]. In addition, others have reported high rates of allelic loss as well as a homozygous deletion in the 5p13-12 region [325, 326]. There has also been cytogenetic and molecular evidence of frequent allele loss of parts of chromosome 11p in lung cancer [327–330]. Apart from allele loss in the 11p13 region, refined mapping of the telomeric 11p15.5 region has suggested the location of two distinct tumor suppressor genes [331]. Loss of genetic material from 11q including the chromosomal region which houses the ATM tumor suppressor gene (11q23) is seen in a number of human cancers including lung [332], breast, ovary, cervix, colon, and skin. Nonetheless, ATM mutations have not been reported in lung cancer.
In addition, the presence of homozygously deleted chromosomal regions 2q33, 8, and X/Y in lung cancer imply yet other unidentified tumor suppressor genes [333]. The comparative genomic hybridization (CGH) technique also detected deletions at 1p, 2q, 3p, 4p, 4q, 5q, 6q, 8p, 9p, 10q, 13q, 17p, 18p, 18q, 21q, and 22q, and characterized the different deletion patterns between SCLC and NSCLC, as well as between adenocarcinoma and squamous cell carcinoma subtypes [11–13, 15].
At 10q23, a candidate tumor suppressor gene, PTEN , is somatically mutated in various tumors including glioblastoma, and prostate, kidney, and breast cancers [334]. PTEN mutations appear infrequent in lung cancers [335] but PTEN expression can be downregulated in lung cancers [336]. PTEN is a part of the PI3K/AKT pathway, and like other examples in lung cancer, other members of this pathway can be involved. Phosphatidylinositol 3-kinase (PI3K) activity is implicated in diverse cellular responses triggered by mammalian cell surface receptors, such as cellular proliferation, growth, apoptosis, and the cytoskeleton, and which are activated in multiple advanced cancers. Unlike other tumors such as colorectal cancers, PIK3CA (encoding the catalytic subunit of PIK3) is only mutated in a small subset of lung cancers [337, 338]. A downstream effector of PI3K is the protein kinase AKT. AKT is negatively regulated by PTEN. AKT appears to be activated in NSCLC lines and seemed to promote survival [339]. Furthermore, nicotine or the tobacco-specific carcinogen, 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone (NNK), could rapidly and potently activate AKT in both NSCLC and SCLC cell lines. Nicotinic activation of AKT increased phosphorylation of multiple downstream AKT substrates including GSK-3, FKHR, tuberin, mTOR, and S6K1 [340].
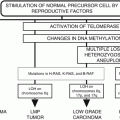
28.5 Molecular Genetic Changes in Preneoplasia
Much of our knowledge about the series of preneoplastic changes in bronchial epithelium has been based on the histological appearances of bronchial epithelial cells. Before the appearance of a clinically overt lung cancer, a sequence of mor phologically distinct changes (hyperplasia, metaplasia, dysplasia and carcinoma in situ) can be observed in bronchial epithelium. The sequential changes for cancers which arise from the proximal airways and bronchi (predominantly squamous cell carcinomas) have long been recognized, whereas changes in peripheral bronchioles and alveoli in evolution to cancers (adenocarcinomas and large cell carcinomas) have been recognized more recently. A number of studies have shown that the preneoplastic cells and bronchial epithelium adjacent to cancers contain a number of genetic abnormalities which are identical to some of the abnormalities found in overt cancer cells. These include abnormalities of MYC and RAS upregulation, cyclin D1 expression, TP53 immunoreactivity, and DNA aneuploidy [10, 260, 341–346]. A follow-up longitudinal study of ex-chromate workers confirmed that some TP53 immuno-positive dysplasias progress to subsequent squamous cell carcinoma [345]. Based on the observation that bronchial dysplastic lesions with elevated telomerase levels, increased Ki-67 index, and expression of TP53 do not regress, it has been suggested that lesions with these molecular changes may progress to invasive squamous carcinomas [347].
In elucidating the temporal sequence of molecular changes, allelotyping analysis of precisely microdissected foci of preneoplastic cells suggests that 3p allele loss is the earliest change followed by 9p allele loss, 17p allele loss (and TP53 mutation), 5q allele loss, and RAS mutations [341, 348–351]. It has been hypothesized that one or more 3p tumor suppressor gene(s) may be critical for proximal squamous lung cancer pathogenesis as abnormalities of 3p appear to be amongst the earliest detectable genetic lesions.
For the adenocarcinoma subtype, based on topographical analysis, it is speculated that KRAS activation may occur early during adenocarcinoma tumorigenesis [346]. In this context, KRAS mutations can be found in atypical adenomatous hyperplasia (AAH), postulated by some to be the precursor lesion for adenocarcinomas [151, 352]. In addition, with laser capture microdissection, EGFR mutations can be demonstrated in apparently morphologically normal appearing bronchial epithelial cells adjacent to tumors known to harbor EGFR mutations [353]. A study of EGFR and KRAS gene mutations in synchronous pulmonary lesions including AAH, carcinomas in-situ (non-mucinous bronchoalveolar carcinoma—BAC), minimally invasive adenocarcinomas, and overtly invasive adenocarcinomas indicated different mutation rates for each gene along this presumed temporal sequence of carcinogenesis [354]. KRAS was mutated in 33 % of AAH, 12 % of carcinomas in situ, 8 % of minimally invasive adenocarcinomas, and 0 % of well-differentiated adenocarcinomas, compared with EGFR mutation rates of 25, 51, 36, 86, and 67 %, respectively. EGFR mutations are found in normal bronchial epithelium and non-malignant epithelium adjacent to tumors in a proportion of patients with AAH and bronchoalveolar carcinoma (BAC), and in 40 % of patients with EGFR mutated adenocarcinomas [353]. In contrast, EGFR mutations are rare in squamous cell carcinomas and therefore EGFR is not implicated in squamous carcinogenesis.
These observations are consistent with the multistep model of carcinogenesis as well as the field cancerization theory proposed by Slaughter. This theory suggests that as the whole aerodigestive tract is repeatedly exposed to carcinogenic damage (tobacco smoke), it is at risk for developing multiple, separate foci of neoplasia [355]. Interestingly, the 3p, 9p, and 17p deletions showed allele-specific loss, defined as deletion of the same allele in preneoplastic tissues as in the primary tumor, even when these lesions were geographically and morphologically distinct [349]. Possible explanations for these allele specific changes include: clonal spread of mutated cells throughout the lung; inherited differences in alleles predisposing to their loss; or some event occurring during lung embryogenesis affecting a particular set of alleles. In support of the clonal spread theory, an identical TP53 point mutation was identified in multiple dysplastic lesions from both lungs of a smoker with chronic obstructive pulmonary disease, thereby indicating an unusual field cancerization mechanism and clonal spread [356].
28.6 Lung Cancer Genome
The combination of the Human Genome Project characterization of human genome sequence and very rapid technological improvements in sequencing tools and bioinformatic analyses has enabled genome-wide investigation of somatic mutations in human cancers. Several large scale sequencing projects have been reported that aim to identify genes that contain driver somatic mutations in tumor samples compared to passenger mutations that do not contribute to carcinogenesis. For instance in one study, more than 1000 somatic mutations were found in 274 megabases (Mb) of DNA corresponding to the coding exons of 518 protein kinase genes in 210 diverse human cancers, of which perhaps 120 genes were thought to harbor driver mutations [357].
In lung cancer, an earlier study used SNP arrays to identify homozygous deletions and chromosome amplifications in primary lung carcinoma and cell lines. Two homozygous deletion regions were identified, one near PTPRD on 9p23 and another in 3q25. High-level amplifications were identified within 8q12-13 in two SCLC specimens, 12p11 in two NSCLC specimens, and 22q11 in four NSCLC specimens. Tyrosine kinase genes which showed high-level amplification included EGFR (3 NSCLC), FGFR1 (2 NSCLC), ERBB2 (1 NSCLC), and MET (1 NSCLC) [31].
Some studies have focused on candidate genes . DNA sequencing of 623 candidate genes in 188 lung adenocarcinomas revealed more than 1000 somatic mutations including in 26 genes that are mutated at significantly high frequencies, such as tyrosine kinase genes; ephrin receptor genes, vascular endothelial growth factor receptor KDR and NTRK genes. These data also provided evidence of somatic mutations in primary lung adenocarcinoma for tumor suppressor genes implicated in other cancers including NF1, APC, RB1, and ATM and for sequence changes in PTPRD as well as the frequently deleted gene LR 1B [358]. Another study searched for somatic mutations in 1507 coding genes from 441 tumors (breast, lung, ovarian, and prostate). These investigators discovered 2576 mutations, with heterogeneity in rates and mutated genes across tumor types and subtypes. The 77 statistical significantly mutated genes included protein kinases, G-protein-coupled receptors such as GRM8, BAI3, AGTRL1, and LPHN3. Another 35 altered genes including GNAS were identified by the integrated evaluation of somatic mutations and copy number alterations [359].
Unbiased studies analyzing the entire genome have recently been reported. Massively parallel sequencing of a SCLC line, NCI-H209 demonstrated 22,910 somatic substitutions, including 134 in coding exons, evidence of different mutation signatures, as well a tandem duplication affecting CHD7 [360]. A comparison of the complete sequences of a primary lung tumor (60× coverage) and adjacent normal tissue (46×) revealed >50,000 high-confidence single nucleotide variants including KRAS [361]. The investigators estimated a 17.7 per megabase genome-wide somatic mutation rate; with a distinct pattern of selection against mutations within expressed genes compared to non-expressed genes and in promoter regions up to 5 kb upstream of all protein-coding genes, as well as a higher rate of amino acid-changing mutations in kinase genes.
Since then, the field is rapidly progressing and an increasing number of NGS studies are now reported, summarized in Table 28.3. Perhaps the most striking findings include identification of novel lung cancer genes, increasing recognition of actionable mutations in adenocarcinoma [362], and emerging molecular themes in cancers arising in different organs [208, 363].
Table 28.3
Next-generation sequen cing studies of lung cancer
First author | Year | Project design | Principal findings |
|---|---|---|---|
Campbell [364] | 2008 | Illumina genome analyzer 2 Lung cancer cell lines WGS | 306 germ line structural changes; 103 somatic rearrangements |
Pleasance [360] | 2010 | SOLiD; 1 SCLC cell line WGS | 22,910 somatic mutations; 134 in coding regions. 58 structural changes. Tobacco associated mutation signatures |
Lee [361] | 2010 | cPAL; 1 NSCLC/normal lung pair WGS | >50,000 SNPs, 392 in coding regions; 43 structural variants |
Ju [365] | 2011 | Illumina HiSeq 2000 and genome analyzer IIx; 1 NSCLC/normal lung pair WGS | 10,724 SNPs, 334 in coding regions. Novel KIF5B-RET fusion |
Imielinski [366] | 2012 | Illumina HiSeq: 183 ACs and matched normal tissue; 159 WES, 23 WES + WGS, 1 WGS | Novel recurrently mutated genes: ARID1A, RBM10, U2AF1. In-frame exonic alterations in EGFR and SIK2 |
The Caner Genome Atlas Research Network [367] | 2012 | Illumina HiSeq: Integrated analysis of 178 SCCs and matched germ line; 19 WGS, 178 WES | Novel mutation in HLA-A; significantly altered pathways: NFE2L2/KEAP1 (34 %), squamous differentiation (44 %), PI3K/AKT (47 %); CDKN2A/RB1 (72 %) |
Peifer | 2012 | Illumina genome analyzer IIx. 29 SCLC exomes, 2 genomes and 15 transcriptomes | High mutation rate: 7.4 ± 1 protein changing mutations per 106; TP53 and RB1 inactivation in all; recurrent mutations in histone modifiers CREBBP, EP300, and MLL |
Govindan [368] | 2012 | Illumina genome analyzer II; 17 WGS; 16AC and 1 large cell carcinoma | Novel mutations in chromatin modification and DNA repair pathways; 14 fusions including ROS, ALK and metabolic enzymes. Possible roles of EGFR and KRAS as tumor initiators |
The Pan-Cancer Project [369] | |||
Ciriello [370] | 2013 | Hierarchical classification of genetic (WES, SNP array, and epigenetic (methylation array) events 3229 tumors of 12 cancer types including 229 lung AC and 182 SCC. | AC are characterized by either mutations or copy number alterations, SCC are characterized by primarily copy number alterations |
Lawrence [371] | 2013 | WGS and WES of 3083 tumors of 12 cancer types including 514 lung cancers: 179 SCC (14 WGS and 165 WES) and 335AC (WES) | Lung AC and SCC have high rates of somatic mutations relative to other tumors: C → A mutations predominate |
Zack [372] | 2013 | Somatic copy number alteration (SCNA) profiling of 357 AC, 344 SCC using SNP array and WGS | Lung AC and SCC have high rates of whole genome duplication relative to other tumor types (59 % AC and 64 % SCC); recurrent focal SCNAs seen in lung SCC and head and neck SCC |
Kandoth [373] | 2013 | Mutational analysis of 3281 tumors from 12 cancer types including 230 lung AC and 178 SCC | The highest mutation rates of all cancers studied were seen in Lung AC and SCC; this was related to TP53 mutations; KEAP1 mutations predominate in lung AC and SCC; EPHA, SETBP1, and STK11 mutations predominate in Lung AC |
Tamborero [374] | 2013 | Mutational analysis of 3205 tumors from 12 tumor types including 226AC and 174 SCC | Lung AC and SCC have high rates of protein activating mutations in high confidence driver mutations relative to other tumor types (median of 9 per tumor) |
Gonzalez-Perez [375] | 2013 | Resequencing data from 4623 exomes from 13 tumor types including 390AC, 31 NSCLC, 174 SCC, 69 SCLC | Int0Gen-mutations platform is a Web based analysis pipeline which is able to summarize genomic data systematically |
Lawrence [376] | 2014 | Predominantly WES of 405AC tumor-normal pairs and 178 SCC tumor-normal pairs | Estimates suggest the number of driver mutations detected in SCC may more than double given sufficient sample size. In excess of 3000 tumor-normal pairs required to detect alterations in 90 % of genes mutated at 2 % above background with 90 % power |
28.6.1 Gene Expression Profiling of Lung Cancer
The expression of regulatory genes of a cell determines its phenotype. Expression can be assessed by measuring its end product, protein, or its intermediate product, mRNA. Microarray technology allows the simultaneous analysis of expression of thousands of genes, generating gene expression patterns that may characterize a disease state. Tumor gene expression data or profiles/signatures can identify unique expression patterns, with implications for predicting cause, i.e., etiology; source, i.e., tissue of origin, and behavior such as prognosis (prognosticator) and response to therapy (predictor). Whilst mRNA levels do not necessarily correlate with protein concentrations in the cell, the efficiency by which mRNA microarrays provide genome wide quantitative information of gene expression data means that expression-based classification of many cancers has now been reported (Table 28.4).
Table 28.4
A selection of gene expression m icroarray studies in lung cancer (N.B. Some studies appear more than once)
Analysis type | Reference | Platforma | No. of probes/elements | Samples in training set (TNM stage)b | Samples in test set (TNM stage)b | Analysis and findingsc |
|---|---|---|---|---|---|---|
Diagnosis/subtype comparison | ||||||
[388] | Affymetrix U95Av2 | 20,951 | 127AC, 21 SCC, 20 carcinoid, 6 SCLC, 12 metastases, 17 NL | – | Unsupervised clustering found histology subclasses | |
[387] | cDNA | >23,000 | 41AC (6 pairs), 16 SCC (5 pairs), 5 LCC, 5 SCLC, 5 NL | – | Unsupervised clustering separated tumors histologically | |
[389] | cDNA | 2400 | 10 SCC, 10AC, 2 SCLC, 5 SCLC cell lines, 1 carcinoid, 2 colon Ca, 1 NL | – | 209 Genes overexpressed in SCLC | |
[395] | Affymetrix HuGeneFL | >7000 | 86AC (I,III), 10 NL | – | Unsupervised clustering found subclasses associated with cancer/non-cancer, stage and differentiation | |
[396] | cDNA | 47,650 | 19AC, 14 SCC, 4 LCC, 2 carcinoid | – | Genes overexpressed in cancer vs non-cancer | |
[397] | cDNA | 1185 | 14AC (I–III), 4 NL | – | DE genes between AC and NL | |
[398] | Oligo | 32 NSCLC | – | Expression profiles correlated with histology | ||
[384] | cDNA | 23,040 | 22AC, 14 SCC, 1 AdSq | – | Unsupervised clustering found subclasses associated with histology and ± lymph node metastasis | |
[399] | cDNA | 425 | 10 NSCLC (I–III) (7AC, 3 SCC) and matched NL | – | DE genes in tumor compared to normal lung. DE genes in stage IA (n = 5) compared to advanced stage (n = 5) | |
[392] | Affymetrix U95Av2 | 20,951 | 21 SCLC cell lines, 18 NL, 8 xenografts | – | Identified DE genes between the two variants of SCLC | |
[400] | cDNA | 5184 | 11 SCC, 9AC, 5 non-SCLC, 3 met LC, 3 NL | 9 NSCLC (4AC, 4 SCC, 1 LCC), 4 met LC | Gene expression predicted tumor (vs. non-tumor), and histology | |
[401] | cDNA | 2400 | 12AC, 3 SCC (I, III) | – | 75 Genes DE between no metastasis, micrometastasis, and overt metastasis | |
[402] | cDNA | 1185 | 13 SCC (I–III), 13AC [397], 4 NL | – | DE genes in SCC and AC compared to NL. DE genes in SCC compared to AC | |
[391] | cDNA | 1185 | – | Identified genes DE between high-grade neuroendocrine carcinomas (SCLC and LCNEC) and other lung cancers | ||
[403] | cDNA | 10,750 | 42 NSCLC (I–III) | – | 62 DE genes between angiogenic compared with non-angiogenic tumors | |
[404] | cDNA | 2305 | 6 SCC, 4 NL | – | 26 DE genes between SCC and NL | |
[52] | Oligo | 10,416 | 79 NSCLC (I–III) (30 pN-, 49 pN+) | 33 NSCLC (I–III) (21 pN0, 12 pN+) | 33-gene signature to predict lymph node metastasis | |
[379] | Affymetrix U133A | >22,000 | 36 Cell lines: 18AC, 4 SCC, 14 SCLC | – | Compared NSCLC to SCLC to identify subtype specific differences. Correlated expression with copy-number differences | |
[405] | Affymetrix U95Av2 and HuGeneFL, cDNA | 2848 | – | Bronchoid, squamoid and magnoid subtypes within AC | ||
[406] | Affymetrix Custom | 59,000 | 89 NSCLC (I–IV) (49 SCC, 40AC), 15 NL | – | 344 DE genes between NSCLC and NL. 72 DE genes between AC and SCC | |
[407] | Oligo | 21,619 | 149 NSCLC (90AC, 35 SCC, 18 LCC, 4 AdSq, 2 LCNEC) | – | 293 DE genes between TRU and non-TRU type AC | |
[390] | cDNA | 32,256 | – | DE in SCLC vs. NL and NSCLC | ||
[408] | Affymetrix U95A and U95Av2 | 20,951 | (1) 48 NSCLC (27 SCC, 21AC), 17 NL; (2) 45 NSCLC (25AC, 20 SCC), 33 NL | (1) 160 NSCLC (139AC, 21 SCC), 6 SCLC, 17 NL; (2) 17 NSCLC (10 SCC, 7AC), 20 NL | 162 DE genes comparing AC, SCC and NL (training set 1). 96 % classification accuracy in 183 samples (test set 1). 20-gene signature (training set 2) had better accuracy (97 %) (test set 2) | |
[409] | cDNA | 7237 | 69 NSCLC (36 SCC, 30AC, 3 LCC) | 75 NSCLC (39AC, 29 SCC, 7 LCC) (IHC validation) | Identified genes correlating with histopathology and EGFR mutation | |
[410] | Affymetrix U133A | >22,000 | 100AC (I–III) | – | Identified clusters that correlated with histological subtypes and KRAS and EGFR mutation status | |
[411] | Affymetrix U133Plus2 | >54,000 | 46 NSCLC (32 SCC, 14AC) and paired NL | 48 NSCLC, 22 NL | Signatures for SCC and AC histology | |
Prognosis | ||||||
[388] | Affymetrix U95Av2 | 20,951 | 127AC, 21 SCC, 20 carcinoid, 6 SCLC, 12 metastases, 17 NL | – | Unsupervised clustering found subgroups with significantly poorer survival | |
[387]
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||||||