Chapter Outline
BASIC CONCEPTS OF FIBRINOLYSIS
COMPONENTS OF THE FIBRINOLYTIC SYSTEM
THE FIBRINOLYTIC ACTIONS OF PLASMIN
PHYSIOLOGIC FUNCTIONS OF THE FIBRINOLYTIC SYSTEM
Wound Healing and Tissue Fibrosis
Atherosclerosis and Cardiovascular Remodeling
Microbial Virulence and Host Defense Mechanisms
Fibrinolysis is the process by which the insoluble protein fibrin is converted to a defined set of soluble degradation products. It occurs in both intravascular and extravascular locations and is essential to human health and survival. Modern molecular biologic techniques have identified the major fibrinolytic genes, the mechanisms regulating their expression, and the consequences of their deficiency or overexpression in genetically engineered mice. In many cases these experiments have yielded surprising and instructive data that have revealed physiologic and pathophysiologic roles for the fibrinolytic system that are much more complex than originally thought. Furthermore, in the current genomic age, molecular analysis of human syndromes has identified specific mutations that result in either thrombosis secondary to fibrinolytic deficiency or hemorrhage secondary to fibrinolytic excess. In this chapter the molecular basis of fibrinolysis and its physiologic roles in human health and disease are reviewed.
Basic Concepts of Fibrinolysis
In response to vascular injury and activation of the coagulation cascade, thrombin induces polymerization of the soluble plasma protein fibrinogen, thereby producing an insoluble, cross-linked end product called fibrin (see Chapter 27 ). Once the flow of blood has been stemmed and vascular integrity restored, fibrin, which is found in both intravascular and extravascular sites, is cleared by a process known as fibrinolysis ( Fig. 28-1, A ). Normally, the fibrinolytic process is tightly regulated by a series of activators, inhibitors, cofactors, and receptors. In the presence of fibrin, which serves as a cofactor for its own destruction, tissue plasminogen activator (tPA) is released from endothelial cells and possibly other cell types and interacts with the circulating zymogen plasminogen. Plasminogen, tPA, and fibrin form a ternary complex that accelerates the catalytic efficiency of plasmin generation by approximately 500-fold. Urokinase is also an efficient plasminogen activator (uPA), but its action is only minimally enhanced by fibrin. The action of plasmin on fibrin generates soluble fibrin degradation products, many of which have their own unique biologic properties.

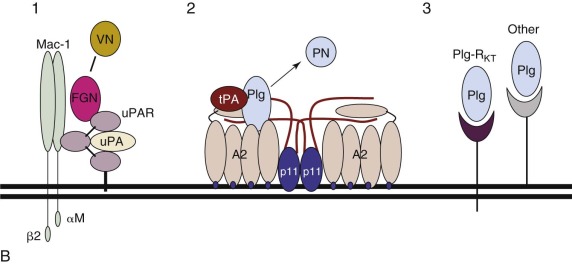
The dynamic regulation of plasmin generation is complex. On the surface of a fibrin-containing thrombus, tPA and plasmin are protected from their major circulating inhibitors plasminogen activator inhibitor 1 (PAI-1) and α 2 -antiplasmin (α 2 -AP), respectively. On release into the circulation, however, plasmin and tPA are rapidly neutralized by these inhibitors and cleared by the liver. Because uPA and the nonenzymatic plasminogen activator streptokinase do not use fibrin as a cofactor, they function well in the fluid phase. Plasminogen may also be activated, albeit rather inefficiently, by proteases of the contact system such as kallikrein, factor XIa, and factor XIIa (see Chapter 27 ).
Cell surfaces represent protected environments in which plasmin can be generated without the risk of neutralization by fluid-phase inhibitors ( Fig. 28-1, B ). Endothelial cells, platelets, monocytes, macrophages, and some tumor cells all express protein receptor sites for plasminogen, tPA, or uPA. The broad substrate specificity of plasmin observed in vitro may relate to its generation in nonvascular sites through fibrin-independent mechanisms. Plasmin may thus play an important role in extravascular events such as the modification of growth and differentiation factors, processing of matrix proteins, and activation of procoagulant molecules.
Components of the Fibrinolytic System
Plasminogen
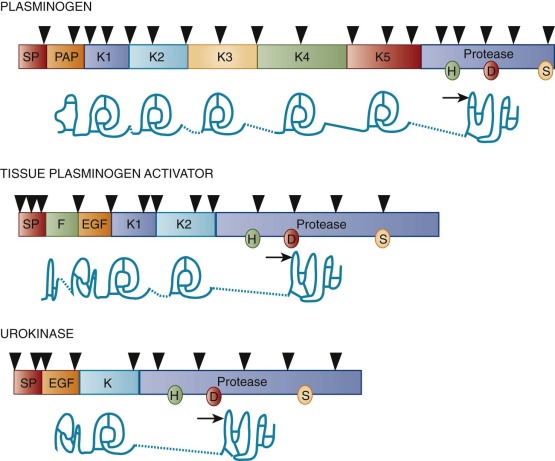
Synthesized primarily in the liver, plasminogen is a single-chain proenzyme with a molecular weight (M r ) of approximately 92,000 that circulates in plasma at a concentration of approximately 1.5 µmol/L ( Table 28-1 ). The plasma half-life of plasminogen in humans is approximately 2 days. Its 791 amino acids are cross-linked by 24 disulfide bridges, 16 of which yield five homologous triple-loop structures called kringles ( Fig. 28-2 ). The first (K1) and fourth (K4) of these 80–amino acid structures with an M r of 10,000 impart high- and low-affinity lysine binding, respectively. The lysine-binding domains of plasminogen appear to mediate its specific interactions with fibrin, cell surface receptors, and other proteins, including its circulating inhibitor α 2 -AP.
| PROTEASES | ||||
| Property | Plasminogen | tPA | uPA | |
| Molecular mass (D) | 92,000 | 72,000 | 54,000 | |
| Amino acids | 791 | 527 | 411 | |
| Chromosome | 6 | 8 | 10 | |
| Site of synthesis | Liver | Endothelium | Endothelium | |
| Neuronal cells | Neuronal cells | Kidney | ||
| Glial cells | Glial cells | |||
| Plasma concentration | ||||
| nmol/L | 1500 | 0.075 | 0.150 | |
| µg/mL | 140 | 0.005 | 0.008 | |
| Plasma half-life | 48 hr | 5 min | 8 min | |
| N -Glycosylation (%) | 2 | 13 | 7 | |
| Form 1 | — | N117, N184, N448 | N302 | |
| Form 2 | N288 | N117, N448 | — | |
| O -Glycosylation | ||||
| α-Fucose | — | T61 | T18 | |
| Complex | T345 | — | — | |
| Two-chain cleavage site | R560-V561 | R275-I276 | K158-I159 | |
| Heavy chain domains | ||||
| Finger | No | Yes | No | |
| Growth factor | No | Yes | Yes | |
| Kringles (no.) | 5 | 2 | 1 | |
| Light-chain catalytic triad | H602, D645, S740 | H322, D371, S478 | H204, D255, S356 | |
| MAJOR SERPIN INHIBITORS | ||||
| Property | α 2 -AP | PAI-1 | PAI-2 | |
| Molecular mass (D) | 70,000 | 52,000 | 60,000 (glycosylated) | |
| 47,000 (nonglycosylated) | ||||
| Amino acids | 452 | 402 | 393 | |
| Chromosome | 18 | 7 | 18 | |
| Sites of synthesis | Kidney, liver | ECs | Placenta | |
| Monocytes/Macrophage | Monocytes/Macrophage | |||
| Hepatocytes | Tumor cells | |||
| Adipocytes | ||||
| Plasma concentration | ||||
| nmol/L | 900 | 0.1–0.4 | ND | |
| µg/mL | 50 | 0.02 | ND | |
| Serpin reactive site | R364-M365 | R346-M347 | R358-T359 | |
| Specificity | Plasmin | uPA = tPA | uPA > tPA | |
| NONSERPIN INHIBITORS | ||||
| Property | TAFI | α 2 -MG | ||
| Molecular mass (D) | 45,000 | 725,000 (monomer ~180,000) | ||
| Amino acids | 423 | 1451 | ||
| Chromosome | 13 | 12 | ||
| Sites of synthesis | Liver | Liver, Macrophage | ||
| Endothelium | ||||
| Fibroblasts | ||||
| Plasma concentration | ||||
| nmol/L | ~75 | ~2000–5000 | ||
| µg/mL | 4 | ~1450–3625 | ||
| Activators | Plasmin >> thrombin | — | ||
| Specificity | C-terminal K and R | Broad spectrum | ||
| SOME PROPOSED ACTIVATION RECEPTORS | ||||
| Property | uPAR | Annexin A2 Complex (A2-p11) 2 | PlgR- KT | |
| A2 | S100A10 (p11) | |||
| Molecular mass (D) | 55-60,000 | 36,000 | 11,000 | 17,000 |
| Amino acids | 313 | 339 | 97 | 147 |
| Chromosome | 19 | 15 | 1 | 9 |
| Source | ECs | ECs | M | Monocytes |
| Monocytes | ||||
| Macrophage | Macrophage | |||
| Fibroblasts | Early myeloid cells | |||
| Tumor cells | Tumor cells | |||
| Ligand(s) | uPA | tPA, Plg | Plg, tPA | Plg |
Posttranslational modification of plasminogen results in two glycosylation variants (forms 1 and 2) (see Table 28-1 ). An O -linked oligosaccharide on Thr345 is common to both forms. Only form 2, however, contains an N -linked oligosaccharide on Asn288. The carbohydrate portion of plasminogen appears to regulate its affinity for cellular receptors and may also specify its physiologic degradation pathway.
Activation of plasminogen results from cleavage of a single Arg-Val peptide bond at position 560 to 561, which produces the active protease plasmin (see Table 28-1 ). Plasmin contains a typical serine protease catalytic triad but exhibits broad substrate specificity in comparison to other proteases of this class. The circulating form of plasminogen, amino-terminal glutamic acid plasminogen (Glu-Plg), is readily converted by limited proteolysis to several modified forms known collectively as lysine plasminogen (Lys-Plg). Hydrolysis of the Lys77-Lys78 peptide bond results in an altered conformation that more readily binds fibrin, displays twofold to threefold higher avidity for cellular receptors, and is activated 10 to 20 times more rapidly than Glu-Plg is. Lys-Plg does not normally circulate in plasma but has been identified on cell surfaces.
Spanning 52.5 kilobases (kb) of DNA on chromosome 6q26-27, the plasminogen gene consists of 19 exons and directs expression of a 2.7-kb messenger RNA (mRNA) (see Fig. 28-2 ). Plasminogen gene activity is stimulated by the acute phase mediator interleukin-6, both in vitro and in vivo. The gene is closely linked and structurally related to that of apolipoprotein(a), an apoprotein associated with the highly atherogenic low-density lipoprotein–like particle lipoprotein(a) and more distantly related to other kringle-containing proteins such as tPA, uPA, hepatocyte growth factor, and macrophage-stimulating protein. The significance of the latter two proteins to the fibrinolytic system remains to be determined.
Plasminogen Activators
Tissue Plasminogen Activator
One of the two major endogenous plasminogen activators, tPA, is a 527–amino acid glycoprotein with an M r of approximately 72,000 (see Table 28-1 ). tPA contains five structural domains, including a fibronectin-like “finger,” an epidermal growth factor–like domain, two kringle structures homologous to those of plasminogen, and a serine protease domain (see Fig. 28-2 ). Cleavage of the Arg275-Ile276 peptide bond by plasmin converts tPA to a disulfide-linked, two-chain form. Although single-chain tPA is less active than two-chain tPA in the fluid phase, both forms demonstrate equivalent activity when bound to fibrin.
The two glycosylation forms of tPA are distinguishable by the presence (type 1) or absence (type 2) of a complex N -linked oligosaccharide moiety on Asn184 (see Table 28-1 ). Both types, however, contain a high-mannose carbohydrate on Asn117, a complex oligosaccharide on Asn448, and an O -linked α-fucose residue on Thr61. The carbohydrate moieties of tPA may modulate its functional activity, regulate its binding to cell surface receptors, and specify degradation pathways.
Located on chromosome 8p12-q11.2, the gene for human tPA is encoded by 14 exons spanning a total of 36.6 kb. Most of the structural domains of tPA are encoded by one or two exons, and the organization of these exons is similar across related domains of tPA and the other fibrinolytic proteases (see Fig. 28-2 ). This observation suggests that the tPA gene arose by an evolutionary process called “exon shuffling,” whereby functionally related genes are generated through rearrangement of exons encoding autonomous domains. Consistent with this hypothesis, various functions of tPA can be localized to specific domains. For example, deletion of the fibronectin-like finger or kringle 2, but not kringle 1, results in a tPA resistant to the cofactor activity of fibrin, whereas catalytic activity in the absence of fibrin remains intact.
The proximal promoter of the human tPA gene contains binding sequences for potentially important transcriptional factors, including AP1, NF1, SP1, and AP2, as well as a potential cyclic adenosine monophosphate (cAMP)-responsive element. In vitro, many agents have been shown to exert small effects on the expression of tPA mRNA, but relatively few enhance tPA synthesis without augmenting PAI-1 synthesis as well. Agents that regulate tPA gene expression independently of PAI-1 include arterial shear stress, thrombin, endotoxin, histamine, butyrate, retinoids, and dexamethasone. Forskolin, which increases intracellular cAMP levels, has been reported to decrease the synthesis of both tPA and PAI-1.
tPA is synthesized in the endothelial cell and stored within Rab3D-negative granules that are distinct from classical Weibel-Palade bodies. Its release is governed by a variety of stimuli such as shear stress, butyrate, thrombin, histamine, bradykinin, epinephrine, acetylcholine, arginine vasopressin, gonadotropins, exercise, and venous occlusion. Its circulating half-life is exceedingly short (≈5 minutes). By itself, tPA is a poor activator of plasminogen. However, in the presence of fibrin, the catalytic efficiency of tPA-dependent plasmin generation ( k cat / K m ) increases by at least 2 orders of magnitude because of a dramatic increase in affinity (decreased Michaelis constant [ K m ]) between tPA and its substrate plasminogen in the presence of fibrin. Although it is expressed by extravascular cells, tPA appears to represent the major intravascular activator of plasminogen.
Urokinase
The second endogenous plasminogen activator, single-chain uPA or prourokinase, is a glycoprotein with an M r of approximately 54,000 and consists of 411 amino acids (see Table 28-1 ). uPA contains an epidermal growth factor–like domain and a single plasminogen-like kringle and possesses a classic catalytic triad within its serine protease domain (see Fig. 28-2 ). Cleavage of the Lys158-Ile159 peptide bond by plasmin or kallikrein converts single-chain uPA to a disulfide-linked two-chain derivative. Located on chromosome 10, the human uPA gene is encoded by 11 exons spanning 6.4 kb and expressed by activated endothelial cells, macrophages, renal epithelial cells, and some tumor cells. As noted earlier, its intron-exon structure is closely related to that of the tPA gene.
uPA may be induced during neoplastic transformation, possibly through a mechanism involving the transcription factors AP1 and AP2. Other agents that appear to induce expression of uPA in vitro include hormones, growth factors, and cAMP. Inflammatory cytokines such as interleukin-1 and lipopolysaccharide induce only small increments in uPA expression, whereas tumor necrosis factor and transforming growth factor β (TGF-β) have a more dramatic (5- to 30-fold) effect.
Two-chain uPA occurs in both high-molecular-weight (M r of 54,000) and low-molecular-weight (M r of 33,000) forms that differ by the presence or absence, respectively, of a 135-residue amino-terminal fragment released by plasmin cleavage between Lys135 and Lys136. Although both forms are capable of activating plasminogen, only the high-molecular-weight form binds to the uPA receptor (uPAR). uPA has much lower affinity for fibrin than tPA does and is an effective plasminogen activator both in the presence and in the absence of fibrin. The extent to which prourokinase possesses intrinsic plasminogen-activating capacity is an area of controversy.
Accessory Plasminogen Activators
Under certain conditions, proteases traditionally classified within the intrinsic arm of the coagulation cascade (see Chapter 26 ) have been shown to be capable of activating plasminogen directly. Such proteases include kallikrein, factor XIa, and factor XIIa. Normally, however, they account for no more than 15% of the total plasmin-generating activity in plasma.
Inhibitors of Fibrinolysis
Plasmin Inhibitors
The action of plasmin is negatively modulated by a family of ser ine p rotease in hibitors called serpins (see Table 28-1 ). All serpins share a common mechanism of action by forming an irreversible complex with the active-site serine of the target protease after proteolytic cleavage of the inhibitor by the target protease. Within such a complex, both protease and inhibitor lose their activity.
A single-chain glycoprotein with an M r of approximately 70,000, α 2 -AP circulates in plasma at relatively high concentrations (≈0.9 µmol/L) and has a plasma half-life of 2.4 days (see Table 28-1 ). This serpin contains about 13% carbohydrate by mass and consists of 452 amino acids with two disulfide bridges. In humans the gene is located on chromosome 18 and contains 10 exons distributed over 16 kb of DNA. The promoter region of the α 2 -AP gene contains a hepatitis B–like enhancer element that directs tissue-specific expression in the liver. α 2 -AP is also a constituent of platelet alpha granules. Plasmin released into flowing blood or in the vicinity of a platelet-rich thrombus is immediately neutralized on forming an irreversible 1 : 1 stoichiometric, lysine binding site–dependent complex with α 2 -AP. Interaction with plasmin is accompanied by cleavage of the Arg364-Met365 peptide bond, and the resulting covalent complexes are cleared in the liver.
Several other proteins inhibit the activity of fibrinolytic serine proteases (see Table 28-1 ). α 2 -Macroglobulin (α 2 -MG) is a tetrameric protein with an M r of 725,000 that is synthesized by the liver, endothelial cells, macrophages, and fibroblasts and is found in platelet alpha granules. The gene for α 2 -MG, which consists of 36 exons distributed over 48 kb of DNA on chromosome 12, directs the expression of a 1451–amino acid polypeptide. As a generic inhibitor of all four classes of proteases (serine, cysteine, aspartyl, and metallo), α 2 -MG is a nonserpin that inhibits plasmin with approximately 10% of the efficiency exhibited by α 2 -AP by forming a noncovalent complex. C1 esterase inhibitor can also serve as an inhibitor of tPA in plasma. Protease nexin may function as a noncirculating cell surface inhibitor of trypsin, thrombin, factor Xa, urokinase, or plasmin and result in protease-inhibitor complexes that are endocytosed via a specific nexin receptor.
Thrombin-activatable fibrinolysis inhibitor (TAFI) is a plasma carboxypeptidase that acts as a potent inhibitor of fibrinolysis (see Table 28-1 ). Identical to the previously cloned carboxypeptidase B and the previously isolated carboxypeptidase U, this single-chain polypeptide with an M r of 45,000 circulates in plasma at concentrations of about 75 nmol/L and undergoes limited proteolysis in the presence of plasmin or thrombin, which leads to its activation. Carboxypeptidase B–like molecules remove carboxyl-terminal lysine or arginine residues from fibrin and other proteins, thereby reducing binding of plasminogen to fibrin and cell surfaces and limiting plasmin generation. The potentially antifibrinolytic effect of thrombin appears to be mediated through its ability to activate TAFI in the presence of thrombomodulin. Anticoagulation by inhibition of factor XI also appears to have an antifibrinolytic effect in vivo by downregulation of thrombin-mediated activation of TAFI. In a system of purified components, TAFI has been shown to reduce tPA-induced fibrinolysis half-maximally at approximately 1 nmol/L, which is well below its concentration in plasma.
Plasminogen Activator Inhibitors
Plasminogen Activator Inhibitor 1.
Of the two major plasminogen activator inhibitors (see Table 28-1 ), PAI-1 is the most ubiquitous. This single-chain cysteine-less glycoprotein with an M r of approximately 52,000 is released by endothelial cells, monocytes, macrophages, hepatocytes, adipocytes, and platelets. Release of PAI-1 is stimulated by many cytokines, growth factors, and lipoproteins common to the global inflammatory response. The PAI-1 gene consists of nine exons spanning 12.2 kb on chromosome 7q21.3-q22. The serpin reactive site is located at Arg346-Met347, and activity of this labile serpin is stabilized upon complex formation with vitronectin, a component of plasma and the pericellular matrix.
Regulation of PAI-1 gene expression is complex. Agents that have been shown to enhance expression of PAI-1 at the message level, the protein level, or both without affecting the synthesis of tPA include the inflammatory cytokines lipopolysaccharide, interleukin-1, tumor necrosis factor α, TGF-β and basic fibroblast growth factor, very-low-density lipoprotein and lipoprotein(a), angiotensin II, thrombin, and phorbol esters. In addition, endothelial cell PAI-1 is downregulated by forskolin and by endothelial cell growth factor in the presence of heparin.
Plasminogen Activator Inhibitor 2.
Originally purified from human placenta, PAI-2 is a 393–amino acid member of the serpin family whose reactive site is the Arg358-Thr359 peptide bond (see Table 28-1 ). The gene encoding PAI-2 is located on chromosome 18q21-23, spans 16.5 kb, and contains eight exons. PAI-2 exists as both a nonglycosylated intracellular form with an M r of 47,000 and a glycosylated form with an M r of 60,000 secreted primarily by macrophages and keratinocytes. Functionally, PAI-2 inhibits both two-chain tPA and two-chain uPA with comparable efficiency (second-order rate constants of 10 5 mmol/L/sec). However, it is less effective in inhibiting single-chain tPA (second-order rate constant of 10 3 mol/L/sec) and does not inhibit prourokinase.
Significant levels of PAI-2 are found in human plasma only during pregnancy but can be markedly enhanced in vitro by the inflammatory mediator tumor necrosis factor. Regulation of PAI-2 gene expression is modulated by a variety of factors, including a number of inflammatory mediators. In macrophages in vitro, secretion of PAI-2 is enhanced by endotoxin and phorbol esters.
Cellular Receptors in Fibrinolysis
Though structurally diverse, cell surface fibrinolytic receptors can be classified into two groups whose integrated actions are likely to be essential for homeostatic control of plasmin activity (see Table 28-1 ). Activation receptors localize and potentiate plasminogen activation, whereas clearance receptors eliminate plasmin and plasminogen activators from the blood or focal microenvironments.
Activation Receptors
Plasminogen Receptors.
Plasminogen receptors are a diverse group of proteins expressed on a wide array of cell types. Receptors reported include α-enolase, glycoprotein IIb-IIIa complex, the Heymann nephritis antigen, amphoterin, and the annexin A2 (A2) complex (A2-p11) (see Table 28-1 ) and have been found to be expressed primarily on monocytoid cells, platelets, renal epithelial cells, neuroblastoma cells, and endothelial cells, respectively. In addition, Plg-R KT , expressed mainly on macrophages, is the first transmembrane plasminogen receptor to be identified, and it appears also to bind tPA (see Table 28-1 ). All plasminogen receptors appear to interact with the kringle structures of plasminogen through carboxyl-terminal lysine residues.
The Urokinase Receptor.
uPAR is expressed on monocytes, macrophages, fibroblasts, endothelial cells, and a variety of tumor cells (see Table 28-1 ). uPAR complementary DNA (cDNA) was cloned and sequenced from a human fibroblast cDNA library and encodes a protein of 313 amino acids with a 21-residue signal peptide. The gene structure consists of seven exons distributed over 23 kb of genomic DNA, which places this glycoprotein within the Ly-1/elapid venom toxin superfamily of cysteine-rich proteins. uPAR is anchored to the plasma membrane through glycosylphosphatidylinositol linkages. uPA bound to its receptor maintains its activity and susceptibility to PAI-1. Formation of uPA–PAI-1 complexes appears to hasten clearance of uPA by hepatic or monocytoid cells.
High-resolution (1.9 to 2.7 Å) analysis of the crystal structure of a complex formed by various amino-terminal uPA peptides and a soluble form of uPAR reveals that uPA engages a central cone-shaped cavity in uPAR formed by its three-finger motif. This arrangement leaves surface residues of uPAR available for binding of other proteins, such as integrins and vitronectin. It is therefore postulated that these interactions may be regulated by binding of uPA itself.
Aside from a potential role in fibrinolysis, uPAR appears to play a pivotal role in cellular signaling and adhesion events. uPAR binds to the adhesive glycoprotein vitronectin at a site distinct from the uPA-binding domain, and uPAR-transfected renal epithelial cells acquire enhanced adhesion to vitronectin and lose their adhesion to fibronectin. Furthermore, uPAR colocalizes with integrins in focal contacts and at the leading edge of migrating cells and also associates with caveolin, a major component of caveolae, structures abundant in endothelial cells and thought to participate in signaling events. Thus integrin function may be regulated by uPAR, an association signifying an integrated relationship between cellular adhesion and proteolysis.
These findings have important implications for the behavior of malignant cells, in which uPAR is frequently overexpressed. High-level uPAR expression also appears to be an indicator of a poor prognosis in breast, lung, colon, esophageal, and gastric cancer. uPA has been identified by protease activity profiling as being important in intravasation of fibrosarcoma, and RNA interference–mediated “knockdown” of uPA and uPAR seems to block the in vivo invasiveness of highly malignant cells.
The Annexin A2 Complex.
Annexin A2 (A2) is a widely distributed, highly conserved, peripheral membrane protein with an M r of 36,000 that is expressed abundantly on endothelial cells, monocyte/macrophages, myeloid cells, and some tumor cells (see Table 28-1 ). It belongs to a more than 60-member superfamily of calcium-dependent, phospholipid-binding proteins that have in common a conserved carboxyl-terminal “core” region preceded by a more variable amino-terminal “tail.” The human ANXA2 gene consists of 13 exons distributed over 40 kb of genomic DNA on chromosome 15 (15q21).
At membrane surfaces, A2 forms a heterotetrameric complex with S100A10 (p11) [(A2-p11) 2 ]. A2 possesses the unique property of binding both plasminogen ( K d of 114 nmol/L) and tPA ( K d of 30 nmol/L), but not uPA, and purified native human A2 stimulates the catalytic efficiency of tPA-dependent plasminogen activation by sixtyfold. This effect is completely inhibited in the presence of lysine analogues or on treatment of A2 with carboxypeptidase B, an agent that removes basic carboxyl-terminal amino acids. Although it lacks a classic signal peptide, A2 is constitutively translocated to the endothelial cell surface within 16 hours of its biosynthesis, where it binds phospholipid via core repeat 2. The (A2-p11) 2 complex may stimulate tPA-dependent plasmin generation even more strongly.
Expression of the A2 complex on the endothelial cell surface appears to be highly regulated. Translocation of A2 to the cell surface requires association with p11, tyrosine phosphorylation of A2 at tyrosine 23 by an src-like kinase, and an activating stimulus such as heat stress or thrombin stimulation. Intracellular concentrations of p11 are regulated by A2, which masks a polyubiquitination site on p11; in the absence of sufficient A2, uncomplexed p11 is rapidly ubiquitinated and directed to the proteasome for degradation. In a negative-feedback mechanism, furthermore, plasmin formed on the endothelial cell surface induces activation of PKC, which phosphorylated serines 11 and 23 on A2, dissociates the (A2-p11) 2 tetramer and reduces further translocation.
Lipoprotein(a), an atherogenic low-density lipoprotein–like particle, competes with plasminogen for binding to A2 and reduces cell surface plasmin generation. Lipoprotein(a) contains an apoprotein called apoprotein(a) that contains kringle structures highly homologous to those of plasminogen. This mechanism may contribute to atherogenesis by reducing fibrinolytic surveillance at the blood vessel wall.
Binding of tPA to A2 requires residues 8 to 13 (LCKLSL) within the amino-terminal “tail” domain of the receptor. This sequence is a target for homocysteine (HC), a thiol-containing amino acid that accumulates in association with nutritional deficiencies of vitamin B 6 , vitamin B 12 , or folic acid or in inherited abnormalities of cystathionine β-synthase, methylenetetrahydrofolate reductase, or methionine synthase; it is associated with atherothrombotic disease. In vitro, HC impairs the intrinsic fibrinolytic system of the endothelial cell by approximately 50% by forming a covalent derivative with Cys9 of A2, thereby preventing its interaction with tPA. The half-maximal dose of HC for inhibition of binding of tPA to A2 is approximately 11 µmol/L HC, a value close to the upper limit of normal for HC in plasma (14 µmol/L).
Studies in homozygous A2-null mice have revealed that A2 contributes to intravascular fibrin balance in vivo ( Table 28-2 ). A2 knockouts display microvascular fibrin deposition, reduced clearance of arterial thrombi, and markedly deficient microvascular endothelial cell surface plasmin generation. In addition, pretreatment of the rat carotid artery with A2 prevents vessel thrombosis in response to injury. A2-null mice also display defects in angiogenesis reflected in several systems, including oxygen-induced retinopathy, a model of retinopathy of prematurity, in which prevention of fibrin formation relieved the block to new blood vessel formation. In mice with diet-induced hyperhomocysteinemia, moreover, fibrin accumulation and impaired angiogenesis were reversed on infusion of fresh, unmodified A2.
| PLASMINOGEN AND FIBRIN | PLASMINOGEN ACTIVATORS | RECEPTORS | INHIBITORS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | A2 Complex | ||||||||||||
| Plg −/− | FgnA −/− | Plg −/− FgnA −/− | tPA −/− | uPA −/− | tPA −/− uPA −/− | ||||||||
| A2 −/− | p11 −/− | uPAR −/− | PAI-1 −/− | PAI-2 −/− | α2AP −/− | TAFI −/− | |||||||
| GENERAL CONSEQUENCES | |||||||||||||
| Development | nl | nl | nl | nl | nl | nl | nl | nl | nl | nl | nl | nl | nl |
| Fertility | ♀↓ | ♀↓ | ♀↓ | nl | nl | ↓ | nl | nl | nl | nl | nl | nl | nl |
| Physical growth | ↓ | nl | nl | nl | nl | ↓ | ↓ | nl | nl | nl | nl | nl | nl |
| Survival | ↓ | ↓/nl | nl | nl | nl | ↓ | nl | nl | nl | nl | nl | nl | nl |
| HEMOSTASIS AND THROMBOSIS | |||||||||||||
| Spontaneous thrombosis | ++ | – | – | – | – | + | + | ND | – | – | – | – | – |
| Induced thrombosis | ND | – | ND | ↑ | ↑ | ↑↑ | ↑ | ND | ND | ↓ | nl | ↓ | nl |
| Lysis of artificial thrombi | ↓ | ND | ND | ↓ | ↓ | ↓↓ | ND | ↓ | nl | ↑ | ND | ↑ | ND |
| Fibrin deposition | ++ | – | – | – | + | ++ | ++ | ++ | – | – | – | – | – |
| Spontaneous hemorrhage | – | ++ | + | – | – | – | – | – | – | – | – | – | – |
In humans, moreover, overexpression of A2 has been associated with hemorrhagic disease. Overexpression of A2 by acute promyelocytic leukemia (APL) cells correlates with the initial severity of hemostatic compromise at the time of diagnosis. Elevated expression of p11 has also been demonstrated in an APL cell line that harbors the [t(15;17)(q22-24;q12-21)] translocation characteristic of APL. It is interesting to note that diet-induced hyperhomocysteinemia has been demonstrated to reverse the hyperfibrinolytic coagulopathy in a murine model of APL.
Conversely, A2 system dysfunction appears to correlate with vascular occlusive disease. Patients with antiphospholipid syndrome and anti-A2 antibodies have a significantly higher risk for clinical thrombosis than do patients who lack this antibody. Similarly, high titer auto-antibodies to A2 are associated with thrombosis and maternal-fetal complications in pregnancy and were unusually prevalent in a cohort of patients with cerebral thrombosis. In sickle cell disease, moreover, A2 polymorphisms are associated with vaso-occlusive stroke, osteonecrosis of bone, and possibly pulmonary hypertension.
Clearance Receptors
Both uPA and tPA are cleared from the circulation via the liver. In vitro, clearance of tPA–PAI-1 complexes also appears to be mediated by a large two-chain receptor called the low-density lipoprotein receptor–related protein (LRP). This complex interaction requires both the growth factor and finger domains of tPA. An additional “receptor-associated protein” with an M r of 39,000 copurifies with LRP and may regulate the binding and uptake of LRP ligands. LRP-knockout embryos undergo developmental arrest by 13.5 days after conception, which suggests that regulation of serine protease activity may be crucial for early embryogenesis. Although several PAI-1–independent clearance pathways for tPA involving the large LRP subunit, the mannose receptor, or an α-fucose–specific receptor have been proposed, in vivo studies in mice suggest that LRP and the mannose receptor play a dominant role in clearance of tPA.
The Fibrinolytic Actions of Plasmin
Fibrinogen
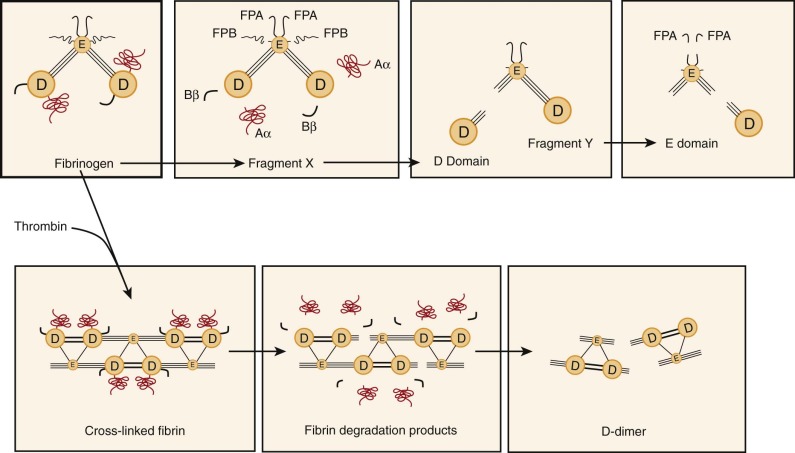
Fibrinogen possesses distinct proteolytic cleavage sites for plasmin and thrombin ( Fig. 28-3 ). Whereas plasmin cleaves carboxyl-terminal Aα and amino-terminal fibrinopeptide B moieties, thrombin primarily releases fibrinopeptide A, which exposes the Gly-Pro-Arg tripeptide sequence and allows fibrinogen to polymerize and form insoluble fibrin (see Chapter 27 ). Cleavage of fibrinogen (M r of 340,000) by plasmin initially produces carboxyl-terminal fragments from the α chain within the D domain of fibrinogen (Aα fragment). Simultaneously, but more slowly, the amino-terminal segments of the β chains are cleaved, with release of a peptide containing fibrinopeptide B. The resulting molecule with an M r of approximately 250,000 is termed fragment X and represents a clottable form of fibrinogen. Additional cleavage events may release the Bβ fragment from the carboxyl-terminal of the β chain, and in a series of subsequent reactions, plasmin cleaves the three polypeptide chains that connect the D and E domains to produce a free D domain (M r of ≈100,000) plus the binodular D-E fragment known as fragment Y (M r of ≈150,000). Finally, domains D and E are separated from each other, and some of the amino-terminal fibrinopeptide A sites on domain E are also modified. Although fragment X can be converted to fibrin by thrombin, the fragments Y, D, and E are all nonclottable and may in fact inhibit the spontaneous polymerization of fibrinogen.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


