Use of new compound such as inhibitors of JAK2 or transforming growth factor β-like molecules might soon revolutionize the treatment of β-thalassemia and related disorders. However, this situation requires careful optimization, noting the potential for off-target immune suppression for JAK2 inhibitors and the lack of mechanistic insights for the use of the ligand trap soluble molecules that sequester ligands of activin receptor IIA and B.
Key points
- •
Increased proliferation of erythroid progenitors is a feature common to many pathologic conditions associated with anemia or an excessive production of red cells.
- •
Chronic stress erythropoiesis (associated with ineffective erythropoiesis and anemia or erythrocytosis) leads to severe comorbidities that aggravate the patient’s condition.
- •
In the context of tumorigenesis, the level of JAK2 activity or activin seems to correlate directly with disease progression.
- •
Development of drugs that target Epo-dependent or Epo-independent pathways, like JAK2 or activin signaling, can be beneficial in hemoglobinopathies and other erythroid disorders.
JAK2 and disorders associated with chronic stress erythropoiesis
Erythropoiesis is a tightly regulated process that includes cytokine signaling and cell-cell interactions in the specialized niche called erythroblastic islands. Because of the essential role of erythrocytes in oxygen delivery, erythropoiesis can effectively respond and adapt quickly to changes in tissue oxygen tension. This situation is mediated primarily by erythropoietin (EPO). EPO signals through the EPO receptor (EPOR). One of the main factors activated by EPO/EPOR interaction is the cytoplasmic kinase JAK2. Through autophosphorylation and crossphosphorylation events, JAK2 activates STAT5 and parallel signaling pathways. STAT5 migrates to the nucleus activating genes necessary for proliferation, differentiation, and survival of erythroid progenitors. The crucial role of EPO, EPOR, JAK2, and STAT5 has been shown by knocking out these factors in mice. Absence of each one of these molecules resulted in a lethal anemia during fetal development. In particular, phosphorylation of STAT5 is essential for basal erythropoiesis and for its acceleration during hypoxic stress (stress erythropoiesis). Some of the conditions in which the JAK2/STA5 pathway is chronically activated and erythropoiesis is accelerated are polycythemia vera and β-thalassemia.
The most recurrent mutation in JAK2, JAK2 V617F, is associated with myeloproliferative neoplasms (MPNs). This mutation is associated with constitutive phosphorylation of JAK2 and EPO hypersensitivity. JAK2 inhibitors such as ruxolitinib, LY2784544, and SAR302503 are currently being tested or used to treat myelofibrosis, essential thrombocythemia, and polycythemia vera. In particular, polycythemia vera, one of the MPNs, is characterized by increased production of erythroid progenitors, erythrocytes, and splenomegaly.
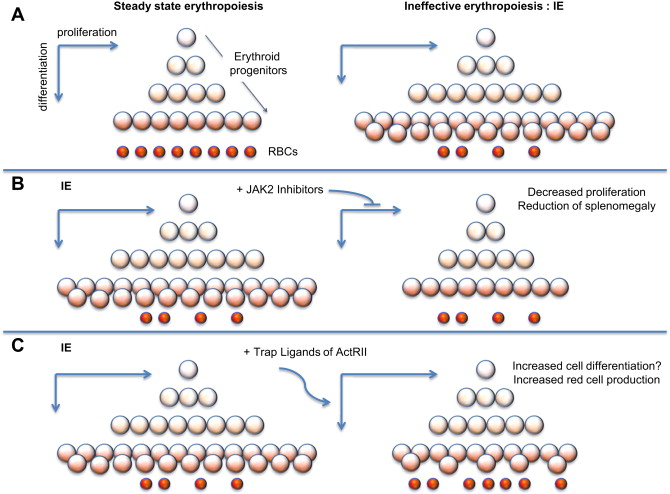
In β-thalassemia, hypoxia leads to high levels of EPO in circulation and, in turn, increased erythroid proliferation. Thus, even in absence of JAK2 mutations, the activity of JAK2 is enhanced, leading to increased proliferation and decreased differentiation of erythroid progenitors (chronic stress erythropoiesis, Fig. 1 A). This situation causes a net increase in the number of erythroid progenitors, leading to extramedullary hematopoiesis and splenomegaly. Splenomegaly, in turn, increases sequestration of red blood cells (RBC), exacerbating the anemia and ineffective erythropoiesis (IE).
JAK2 and disorders associated with chronic stress erythropoiesis
Erythropoiesis is a tightly regulated process that includes cytokine signaling and cell-cell interactions in the specialized niche called erythroblastic islands. Because of the essential role of erythrocytes in oxygen delivery, erythropoiesis can effectively respond and adapt quickly to changes in tissue oxygen tension. This situation is mediated primarily by erythropoietin (EPO). EPO signals through the EPO receptor (EPOR). One of the main factors activated by EPO/EPOR interaction is the cytoplasmic kinase JAK2. Through autophosphorylation and crossphosphorylation events, JAK2 activates STAT5 and parallel signaling pathways. STAT5 migrates to the nucleus activating genes necessary for proliferation, differentiation, and survival of erythroid progenitors. The crucial role of EPO, EPOR, JAK2, and STAT5 has been shown by knocking out these factors in mice. Absence of each one of these molecules resulted in a lethal anemia during fetal development. In particular, phosphorylation of STAT5 is essential for basal erythropoiesis and for its acceleration during hypoxic stress (stress erythropoiesis). Some of the conditions in which the JAK2/STA5 pathway is chronically activated and erythropoiesis is accelerated are polycythemia vera and β-thalassemia.
The most recurrent mutation in JAK2, JAK2 V617F, is associated with myeloproliferative neoplasms (MPNs). This mutation is associated with constitutive phosphorylation of JAK2 and EPO hypersensitivity. JAK2 inhibitors such as ruxolitinib, LY2784544, and SAR302503 are currently being tested or used to treat myelofibrosis, essential thrombocythemia, and polycythemia vera. In particular, polycythemia vera, one of the MPNs, is characterized by increased production of erythroid progenitors, erythrocytes, and splenomegaly.
In β-thalassemia, hypoxia leads to high levels of EPO in circulation and, in turn, increased erythroid proliferation. Thus, even in absence of JAK2 mutations, the activity of JAK2 is enhanced, leading to increased proliferation and decreased differentiation of erythroid progenitors (chronic stress erythropoiesis, Fig. 1 A). This situation causes a net increase in the number of erythroid progenitors, leading to extramedullary hematopoiesis and splenomegaly. Splenomegaly, in turn, increases sequestration of red blood cells (RBC), exacerbating the anemia and ineffective erythropoiesis (IE).
Potential use of JAK2 inhibitors in β-thalassemia
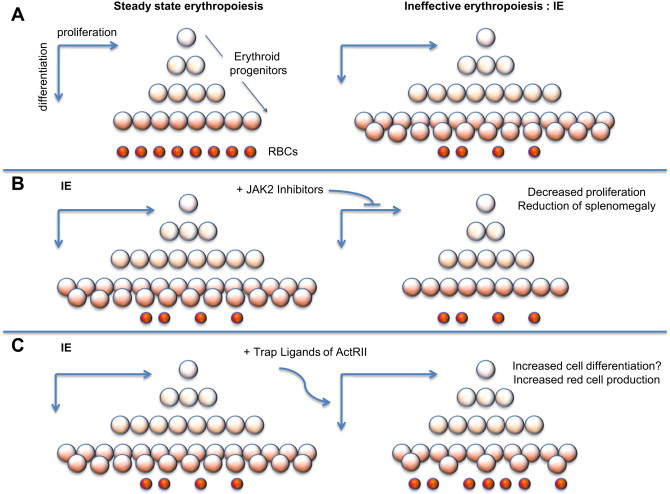
IE is the hallmark of a group of anemias characterized by cell death associated with increased proliferation and decreased differentiation of erythroblasts, leading to an expansion in the number of erythroid progenitors, but with suboptimal or absent production of normal RBC. The discovery that JAK2 plays an important role in the progression and exacerbation of IE suggests that drugs inhibiting the activity of JAK2 could mitigate IE (see Fig. 1 B) and reverse splenomegaly. In preclinical studies, it has been shown that a JAK2 inhibitor (200mg/Kg/d dose) dramatically decreased the spleen size and modulated the IE. Based on these observations, use of JAK2 inhibitors could be used to reverse splenomegaly, thereby avoiding the need for splenectomy. Ideally, this treatment could also be helpful in reducing the rate of blood transfusions. leading to improved management of anemia and iron overload.
Activins, members of the transforming growth factor β family signaling
Activins are soluble ligands, which, along with bone morphogenetic proteins (BMPs) and growth and differentiation factors (GDFs), belong to a large group of proteins called the transforming growth factor β (TGF-β) family. The most well-characterized activins are homodimeric or heterodimeric structures composed of 2 similar β-chains, A or B. Inhibins, on the other hand, are heterodimers composed of α-chains and β-chains (from activins) and antagonize activins and BMP signaling. Generally, ligands of TGF-β family are synthesized from a common precursor with a prodomain that can determine the activity and localization of a ligand.
The TGF-β family signal through 7 different type I and 5 different type II transmembrane serine/threonine kinase receptors. Type I receptors (activin receptorlike kinases) ALK2, ALK4, and ALK7 (also indicated as ACVR1, ACVR1B, and ACVR1C, respectively) and activin receptor IIA (ActRIIA) and ActRIIB (also indicated as ACVR2A, ACVR2B) are typically the mediators of activin effect. ActRIIs are shared by some of the BMPs and GDFs. Activin signaling is carried out through formation of a ternary complex between the ligand, the type II and the type I receptors, which phosphorylates SMAD proteins. SMADs multimerize, and these complexes translocate to the nucleus and regulate gene expression in concert with other transcription factors. Follistatin (FST) and FST-related protein (FRP) bind extracellularly to activins and other related TGF-β ligands, controlling their signaling and availability. FST inhibits activin by hiding one-third of its residues as well as type I and II receptor binding sites. Mice with a disrupted FST gene have musculoskeletal and skin abnormalities, whereas mice with the gene encoding FRP (FST-related gene or FLRG) deleted show dysregulated glucose metabolism and fat homeostasis.
Activins are expressed in various tissues and have a broad range of activities that regulate:
- •
Gonadal function
- •
Hormonal homeostasis
- •
Growth and differentiation of musculoskeletal tissues
- •
Growth and metastasis of cancer cells
- •
Proliferation and differentiation of embryonic/hematopoietic stem and erythropoietic cells
- •
Higher brain function
Activin activities are involved in the cause and pathogenesis of several diseases. Dysregulation of activin signaling has been associated with many malignant disorders as well as diseases affected by anemia. For these conditions, the inhibition of activin signaling represents an interesting therapeutic approach.
Cancer-related anemia and ineffective erythropoiesis
Anemia is a condition that affects hematologic malignancies like multiple myeloma and myelodysplastic syndromes (MDS). MDS encompass a heterogeneous group of closely related clonal hematopoietic disorders characterized by a marrow with impaired maturation (dysmyelopoiesis) and peripheral blood cytopenias, resulting from ineffective blood cell production and develop when a clonal mutation predominates in the bone marrow, suppressing healthy stem cells. Anemia can also be caused by myelosuppressive chemotherapy. Anemia is also observed in some tumors in absence of chemotherapy treatment and it can be considered a prognostic factor of reduced survival. IE also leads to anemia and it is the hallmark of a group of diseases such as β-thalassemia. Individuals with these anemias have markedly increased EPO levels, which result in massive erythroid expansion in the hypercellular marrow. When abnormalities in the red cells lead to their intramedullary demise, erythropoiesis is ineffective and leads to enhanced intestinal iron absorption, and resultant tissue iron loading and toxicity. The thalassemia syndromes (both α-thalassemia and β-thalassemia) are the most frequent conditions associated with IE and result from diminished production of α-globin or β-globin chains, respectively, the 2 subunits of the hemoglobin A (adult) molecule. As a result of IE, patients develop several comorbidities, including iron overload and bone abnormalities. Another group of conditions characterized by IE are congenital and acquired sideroblastic anemias. In these conditions, the bone marrow produces sideroblasts, characterized by granules of iron accumulated in perinuclear mitochondria, forming a ring around the erythroblast nucleus. These ringed sideroblasts fail to differentiate into healthy erythrocytes. Acquired sideroblastic anemias may be caused either by a genetic disorder or indirectly as part of the MDS. Other erythroid disorders that might benefit from ameliorating the IE are congenital dyserythropoietic anemias, chronic pernicious anemia, and hereditary spherocytosis.
Individuals affected by thalassemia and MDS receive blood transfusion to compensate for the anemia, but with time, several comorbidities develop, including iron overload, bone abnormalities, and, in patients with MDS, progression to acute myeloid leukemia. Although improved transfusion and iron chelation treatments over the past 2 decades or so have reduced morbidity and improved the life expectancy of patients, they do not provide a definitive cure and lack the ability to correct IE. Therefore, alternative pharmacologic therapies that target the direct recovery of terminal erythroid differentiation are urgently needed.
The use of EPO and its derivatives, also referred to as erythropoiesis stimulating agents (ESAs), to treat cancer-related anemia can be controversial, because it has been speculated that it might aggravate tumor progression. In addition, ESAs have been used for diseases characterized by anemia caused by IE but with limited benefits. Therefore, new pharmacologic treatments with different mechanisms of action are needed. In this perspective, the use of molecules that can target EPO-unrelated pathways, like activin signaling, might be potential candidates to ameliorate anemia.
Effect of activin signaling in bone
In vitro and in vivo data on activin signaling are discordant. Mice injected with activin A show increase of bone formation and bone strength, although in rats, similar results were reproduced using inhibin A. In perimenopausal and postmenopausal women, follicle-stimulating hormone (FSH) seems to indirectly exert its anabolic effect on bone through an ovarian mediator (possibly inhibin A). Withdrawal of inhibin A could be related to bone loss observed in perimenopause and postmenopause. FST has been shown to have an opposite effect, indicating that FST and inhibins do not operate as analogue in the bone context. In relationship to metastatic bone progression, increased levels of circulating activin A seem to be a prognostic factor. Patients who have breast and prostatic cancer with bone metastasis present higher levels of activin A than nonmetastatic patients. A similar observation was made for patients with multiple myeloma, in whom high activin levels correlate with extensive bone involvement and lower survival rate. Moreover, in vitro studies have shown that multiple myeloma cells stimulate stromal cells to produce activin, which in turn leads to inhibition of osteoblast differentiation in vitro.
Effect of activin signaling in cancer
In vivo studies showed that lack of inhibin in transgenic mice causes gonadal and adrenal tumorigenesis, indicating that these activin repressors are important tumor repressors in these tissues. Overexpression of FST in these mice does not reduce tumor incidence but modulates tumor progression and reduces the tumor cachexia-like syndrome associated with high levels of activin. As for the liver, FST adenoviral-induced overexpression causes hyperproliferation of hepatocytes. The opposite effect is observed when overexpression of FST is induced in small cell lung cancer cells, which seem to produce a reduction of experimental metastases in various organs in NOD-SCID (nonobese diabetic/severe combined immunodeficiency) mice. Compared with transgenic activin-deficient mice, FST-deficient mice die before birth because of compromised development of growth, skin, muscle, and skeletal development. Therefore, FST operates on many signaling pathway in addition to the activin-related ones. The function of FST and activin in different tissues can differ vastly, and therefore, their levels as indicators of tumor development, progression, and metastases might be challenging, although certainly impactful.
Effect of activin signaling in hematopoiesis and erythropoiesis
Members of the TGF-β family are also key regulators of human hematopoiesis, modulating various cellular responses, such as proliferation, differentiation, migration, and apoptosis. Activin expression can be detected in bone marrow cells, including erythroid cells. Recombinant activin induces cellular shrinking and nuclear condensation of CD34 + cells during in vitro differentiation. This process is reverted by addition of FRP, BMP2, and BMP4.
Activin A and BMP2 and BMP4, alone or in combination, have been shown to have a role in the regulation of erythropoiesis in various models.
The biological function of activin A, BMP2, and BMP4 was assessed measuring clonogenic potential in colony forming cell assay of human CD34 + cells isolated from either mobilized peripheral blood or bone marrow. Activin was found to increase the number of both late erythroid burst-forming unit (BFU-E) and erythroid colony-forming unit (CFU-E). This observation was confirmed in vivo through injection of activin in anemic (phlebotomized) and normal mice. On the other hand, BMP2 was found to increase the number of early BFU-E. Cells treated with activin A showed a significant decrease in nuclear size, a phenomenon associated with maturation of erythroid cells. Compared with activin A alone, addition of BMP4 induces an inhibitory effect in the number of late BFU-E and CFU-E and increases nuclear size. This finding suggests that BMP4 facilitates differentiation of the erythroid progenitors before they lose their ability to form colonies and their nuclear size starts shrinking. BMP2 antagonizes the effect of activin A on nuclear size reduction. Therefore, BMP molecules may have a different effect on the activin A–mediated biological activities.
In hematopoiesis, FST has an inhibitory effect on activin. Both FST and FRP also neutralize GDF11 and myostatin. Both repressors interfere with the ability of activin to induce nuclear size reduction and increase counts of early BFU and CFU, and also inhibit BMP2-induced early BFU proliferation. Bone morphogenetic stromal cells are among the tissues that produce at steady state the highest levels of FLRG transcripts. They also produce activin and FST. Among hematopoietic lineages, monocytes have the highest level of expression of FLRG. Expression of FLRG in erythroid cells is mostly observed in immature cells, whereas FST is expressed at highest levels in most mature erythroid cells. The expression of both FST and FLRG transcripts are induced by activin A as well as TGF-β, indicating that these 2 repressors participate in a negative feedback loop that regulates activin signaling. Additional regulation of activin and TGF-β ligand signaling pathways occurs intracellularly via the SMAD inhibitory proteins 6 and 7. These proteins induce ubiquitin-dependent degradation of SMAD2, 3, and 4.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


