In this article, the authors discuss new approaches to treating iron overload diseases using hepcidin mimetics or by modulating endogenous hepcidin expression. In particular, the authors discuss lipid nanoparticle encapsulated siRNA and antisense oligonucleotide–mediated inhibition of TMPRSS6, an upstream regulator of hepcidin, and treatment with transferrin or hepcidin mimetics, including the recently described minihepcidins . In each case, in animal models of β-thalassemia, not only do the interventions affect iron absorption but they also act as disease-modifying agents that ameliorate the ineffective erythropoiesis.
Key points
- •
Dysregulation of iron metabolism is a primary or secondary cause of morbidity and mortality in many diverse diseases, including hereditary hemochromatosis and β-thalassemia.
- •
Hepcidin is the central hormonal regulator of iron metabolism.
- •
Tmprss6 is a serine protease that regulates hepcidin expression by the hepatocyte through a mechanism that involves several of the hereditary hemochromatosis proteins.
- •
Modulation of hepcidin activity has demonstrated potential as a treatment modality to treat iron overload disorders in preclinical animal models.
Introduction to iron metabolism
Because iron is highly toxic when present in excess, mammals have evolved elaborate mechanisms for the regulation of iron acquisition, transport, storage, and utilization. A typical adult human is endowed with approximately 4 g of iron, almost two-thirds of which is distributed in hemoglobin in red blood cells (RBCs). Nearly 25 mg of iron is required to support erythropoiesis each day, but most of the iron required for erythropoiesis derives from recycling of iron from effete RBCs by macrophages of the reticuloendothelial system. Under normal conditions, only 1 to 2 mg of iron is absorbed each day from the diet; that is only to offset iron losses, which are not regulated, and limited to physiologic and nonphysiologic epithelial cell (eg, skin and intestine) or blood loss. Accordingly, total body iron is regulated entirely at the level of intestinal absorption, which can be modulated according to the body’s needs.
The Hepcidin-Ferroportin Iron Regulatory Axis
Hepcidin is a peptide hormone produced predominately by the liver in response to iron stores. As iron levels increase, so does hepcidin, which, as a negative regulator of iron release from cells, binds to and causes the internalization and degradation of ferroportin (FPN1), the only known iron exporter. FPN1 is expressed in abundance on macrophages and duodenal enterocytes, the cells that are directly responsible for iron recycling from senescent RBCs and for iron absorption from the intestine ( Fig. 1 ). Thus, hepcidin production simultaneously leads to decreased intestinal iron absorption and sequestration of iron in macrophages, limiting its availability for erythropoiesis. Conversely, decreasing hepcidin expression permits more nonheme iron to be taken up from the diet and released from internal stores. A failure of this stores regulator of systemic iron metabolism underlies the pathophysiology of most forms of hereditary hemochromatosis (HH) (see later discussion).
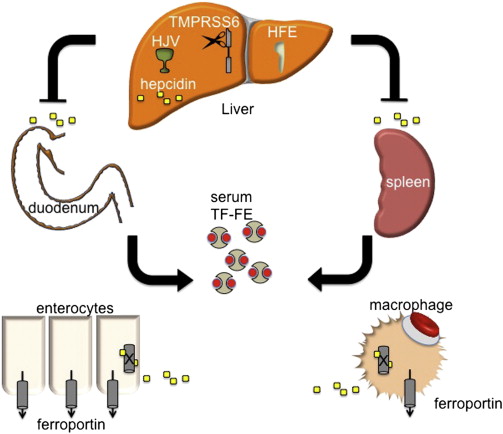
In addition to systemic iron deficiency, hepcidin expression is also suppressed by anemia and hypoxia. Anemias characterized by ineffective erythropoiesis (bone marrow erythroid hyperplasia with premature, intramedullary death of maturing erythroblasts) seem to uniquely potently suppress hepcidin production even in the presence of systemic iron overload . The factor or factors that communicate this signal from the bone marrow to the liver to suppress hepcidin have been termed the erythroid regulator of iron metabolism. It is the apparent supremacy of the erythroid regulator compared with the stores regulator that underlies the pathogenesis of iron overload in iron-loading anemias , such as β-thalassemia intermedia, which are characterized by ineffective erythropoiesis. Importantly, in these anemias, as well as in HH, the regulatory dysfunction leading to iron overload is a relative if not absolute deficiency in hepcidin for the degree of iron overload. It is on this theoretical basis that upregulation of hepcidin has been envisioned as a means to treat iron overload in these apparently diverse diseases.
Iron-Responsive Hepcidin Expression by the Hepatocyte
It is now evident that the autosomal recessive forms of HH caused by mutations in HFE , HJV , or TFR2 result from a disruption of the hepatocyte’s ability to translate systemic iron stores and availability for erythropoiesis represented by the transferrin saturation (or concentration of diferric transferrin) into a signal that promotes hepcidin gene transcription. In this way, they are thought to disrupt the stores regulator of systemic iron homeostasis. This pathway has been reviewed comprehensively elsewhere. Only elements that are fundamental to the therapeutic innovations discussed later are highlighted here.
There is strong evidence that the bone morphogenetic protein (BMP)–sons of mothers against decapentaplegic (SMAD) signaling pathway plays a key role in the regulation of hepcidin and systemic iron metabolism ( Fig. 2 ). Hemojuvelin (HJV), which is mutated in patients with a severe, juvenile onset from of HH, is a BMP coreceptor protein that facilitates signaling through the BMP type I receptors (BMPRIs) ALK2 and ALK3 in response to BMP6, which is itself upregulated in the liver by iron. Activated BMP receptors phosphorylate SMADS1, 5, and 8, which in turn phosphorylate SMAD4, which translocates to the nucleus, stimulating transcription by binding to a BMP-response element in the hepcidin promoter.
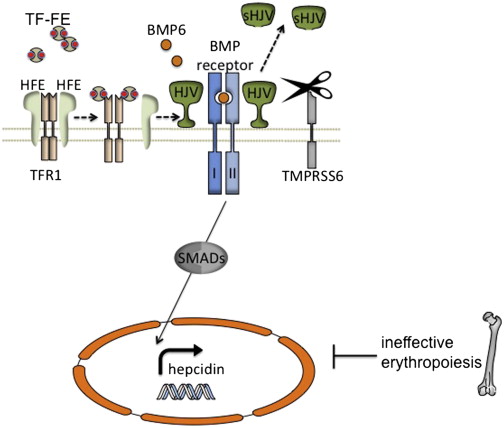
Although juvenile hemochromatosis is rare, mutations in the classic hereditary hemochromatosis gene (HFE) account for most of the patients with HH in the Western hemisphere. Early work demonstrated that HFE interacts with the transferrin receptor (TFRC or TFR1) in a manner that can be competitively inhibited by diferric transferrin binding to TFR1. A second transferrin receptor, TFR2, mutated in a fraction of patients with HH, also interacts with HFE in vitro, but it associates with diferric transferrin only very poorly. In this way, there would seem to be a means for hepatocytes to sense the amount of iron in the plasma. It is a matter of debate whether TFR2, HFE, or both directly interact with the HJV-BMPR complex ; however, inactivation of either protein diminishes the activation of the downstream SMADS as well as hepcidin transcription in response to iron.
Mutations in any of the HH proteins or certain components of the BMP-SMAD signaling cascade in hepatocytes in model organisms lead to hyporesponsiveness of hepcidin transcription in response to iron. In contrast, mutations in TMPRSS6, a membrane associated protease expressed solely in the liver, cause the opposite phenotype: excessively high hepcidin levels in response to a given iron status, and the clinical phenotype of congenital iron deficiency. Work in vitro suggests that TMPRSS6 regulates HJV protein levels at the cell membrane by cleaving it to generate a soluble form (see Fig. 2 ). It seems that iron, BMP6, and hypoxia are all able to induce TMPRSS6 transcription in vivo. In toto, TMPRSS6 activity and expression are coordinated to inhibit hepcidin transcription mediated by the BMP-SMAD pathway and in doing so may ordinarily help to prevent excessive hepcidin induction and prevent iron deficiency.
Introduction to iron metabolism
Because iron is highly toxic when present in excess, mammals have evolved elaborate mechanisms for the regulation of iron acquisition, transport, storage, and utilization. A typical adult human is endowed with approximately 4 g of iron, almost two-thirds of which is distributed in hemoglobin in red blood cells (RBCs). Nearly 25 mg of iron is required to support erythropoiesis each day, but most of the iron required for erythropoiesis derives from recycling of iron from effete RBCs by macrophages of the reticuloendothelial system. Under normal conditions, only 1 to 2 mg of iron is absorbed each day from the diet; that is only to offset iron losses, which are not regulated, and limited to physiologic and nonphysiologic epithelial cell (eg, skin and intestine) or blood loss. Accordingly, total body iron is regulated entirely at the level of intestinal absorption, which can be modulated according to the body’s needs.
The Hepcidin-Ferroportin Iron Regulatory Axis
Hepcidin is a peptide hormone produced predominately by the liver in response to iron stores. As iron levels increase, so does hepcidin, which, as a negative regulator of iron release from cells, binds to and causes the internalization and degradation of ferroportin (FPN1), the only known iron exporter. FPN1 is expressed in abundance on macrophages and duodenal enterocytes, the cells that are directly responsible for iron recycling from senescent RBCs and for iron absorption from the intestine ( Fig. 1 ). Thus, hepcidin production simultaneously leads to decreased intestinal iron absorption and sequestration of iron in macrophages, limiting its availability for erythropoiesis. Conversely, decreasing hepcidin expression permits more nonheme iron to be taken up from the diet and released from internal stores. A failure of this stores regulator of systemic iron metabolism underlies the pathophysiology of most forms of hereditary hemochromatosis (HH) (see later discussion).
In addition to systemic iron deficiency, hepcidin expression is also suppressed by anemia and hypoxia. Anemias characterized by ineffective erythropoiesis (bone marrow erythroid hyperplasia with premature, intramedullary death of maturing erythroblasts) seem to uniquely potently suppress hepcidin production even in the presence of systemic iron overload . The factor or factors that communicate this signal from the bone marrow to the liver to suppress hepcidin have been termed the erythroid regulator of iron metabolism. It is the apparent supremacy of the erythroid regulator compared with the stores regulator that underlies the pathogenesis of iron overload in iron-loading anemias , such as β-thalassemia intermedia, which are characterized by ineffective erythropoiesis. Importantly, in these anemias, as well as in HH, the regulatory dysfunction leading to iron overload is a relative if not absolute deficiency in hepcidin for the degree of iron overload. It is on this theoretical basis that upregulation of hepcidin has been envisioned as a means to treat iron overload in these apparently diverse diseases.
Iron-Responsive Hepcidin Expression by the Hepatocyte
It is now evident that the autosomal recessive forms of HH caused by mutations in HFE , HJV , or TFR2 result from a disruption of the hepatocyte’s ability to translate systemic iron stores and availability for erythropoiesis represented by the transferrin saturation (or concentration of diferric transferrin) into a signal that promotes hepcidin gene transcription. In this way, they are thought to disrupt the stores regulator of systemic iron homeostasis. This pathway has been reviewed comprehensively elsewhere. Only elements that are fundamental to the therapeutic innovations discussed later are highlighted here.
There is strong evidence that the bone morphogenetic protein (BMP)–sons of mothers against decapentaplegic (SMAD) signaling pathway plays a key role in the regulation of hepcidin and systemic iron metabolism ( Fig. 2 ). Hemojuvelin (HJV), which is mutated in patients with a severe, juvenile onset from of HH, is a BMP coreceptor protein that facilitates signaling through the BMP type I receptors (BMPRIs) ALK2 and ALK3 in response to BMP6, which is itself upregulated in the liver by iron. Activated BMP receptors phosphorylate SMADS1, 5, and 8, which in turn phosphorylate SMAD4, which translocates to the nucleus, stimulating transcription by binding to a BMP-response element in the hepcidin promoter.
Although juvenile hemochromatosis is rare, mutations in the classic hereditary hemochromatosis gene (HFE) account for most of the patients with HH in the Western hemisphere. Early work demonstrated that HFE interacts with the transferrin receptor (TFRC or TFR1) in a manner that can be competitively inhibited by diferric transferrin binding to TFR1. A second transferrin receptor, TFR2, mutated in a fraction of patients with HH, also interacts with HFE in vitro, but it associates with diferric transferrin only very poorly. In this way, there would seem to be a means for hepatocytes to sense the amount of iron in the plasma. It is a matter of debate whether TFR2, HFE, or both directly interact with the HJV-BMPR complex ; however, inactivation of either protein diminishes the activation of the downstream SMADS as well as hepcidin transcription in response to iron.
Mutations in any of the HH proteins or certain components of the BMP-SMAD signaling cascade in hepatocytes in model organisms lead to hyporesponsiveness of hepcidin transcription in response to iron. In contrast, mutations in TMPRSS6, a membrane associated protease expressed solely in the liver, cause the opposite phenotype: excessively high hepcidin levels in response to a given iron status, and the clinical phenotype of congenital iron deficiency. Work in vitro suggests that TMPRSS6 regulates HJV protein levels at the cell membrane by cleaving it to generate a soluble form (see Fig. 2 ). It seems that iron, BMP6, and hypoxia are all able to induce TMPRSS6 transcription in vivo. In toto, TMPRSS6 activity and expression are coordinated to inhibit hepcidin transcription mediated by the BMP-SMAD pathway and in doing so may ordinarily help to prevent excessive hepcidin induction and prevent iron deficiency.
Preclinical investigation of hepcidin mimetic and hepcidin-induction therapies in murine models of HH
As described earlier, because of the relative hepcidin deficiency seen in both HH and in the iron-loading anemias associated with ineffective erythropoiesis, manipulation of hepcidin levels either through exogenous administration or endogenous stimulation has been envisioned as a potential pharmacologic approach to these disorders ( Box 1 ). In many ways, because of the safety, efficacy, and low cost of phlebotomy for the treatment of HH, many have seen using animal models of HH as the proof of principle for these therapies as a prelude to the more complicated secondary hepcidin suppression seen in the iron-loading anemias.
| Genetic ablation of Tmprss6 ( Tmprss6 -/- ) | Murine β-thalassemia intermedia ( Hbb th3/+ ), murine HH ( Hfe -/- ) |
| Transferrin ( Tf ) therapy | Murine β-thalassemia intermedia ( Hbb th1/th1 ) |
| Transgenic overexpression of hepcidin ( Hamp1 ) | Murine β-thalassemia intermedia ( Hbb th3/+ ) and murine HH ( Hfe -/- ) |
| Dietary iron restriction | Murine β-thalassemia intermedia ( Hbb th3/+ ) |
| Pharmacologic repression of Tmprss6 expression (siRNA, antisense oligonucleotide) | Murine β-thalassemia intermedia ( Hbb th3/+ ), murine HH ( Hfe -/- ) |
| Treatment with hepcidin mimetic (minihepcidin) | Murine juvenile HH ( Hamp1 -/- ) |
| BMP6 therapy | Murine HH ( Hfe -/- ) |
Transgenic Hepcidin Overexpression in HFE HH
Hepcidin therapy was first attempted by Nicolas and colleagues in a murine model of HH. This group showed that Hfe -/- animals have inappropriately low hepcidin expression that does not change as the animals age and load iron ( Fig. 3 A ). Earlier work in this laboratory had generated a transgenic animal that overexpressed the mouse hepcidin gene under control of the liver-specific transthyretin promoter. These transgenic animals are extremely pale; have diminished whole body iron stores and a severe hypochromic, microcytic anemia; and, on a C57BL/6 background, die within a few hours of birth. Transgenic mice on a mixed 129Sv-C57BL/6 background are viable, and the anemia eventually subsides as the endogenous hepcidin normalizes at approximately 9 weeks of life. More severely affected founder animals require treatment with exogenous iron to survive past weaning. As one would expect, overexpression of hepcidin in Hfe -/- animals (see Fig. 3 B) greatly decreased iron loading in whole embryos and also in the livers of 1- and 2-month-old HH animals. In fact, in many animals, the correction of the hepcidin deficiency was too great, leading to iron deficiency anemia. In toto, this work was the proof in principle for hepcidin therapy in iron overload diseases, but it equally illustrated the potential complications of an inability to titer the therapy to the physiologic state of the animal.
Related posts:
 Ischemia-reperfusion Injury in Sickle Cell Anemia
Ischemia-reperfusion Injury in Sickle Cell Anemia
 Gene Therapy for Hemoglobinopathies
Cellular Adhesion and the Endothelium
Hemoglobin S Polymerization and Red Cell Membrane Changes
Gene Therapy for Hemoglobinopathies
Cellular Adhesion and the Endothelium
Hemoglobin S Polymerization and Red Cell Membrane Changes
 Inflammatory Mediators of Endothelial Injury in Sickle Cell Disease
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Inflammatory Mediators of Endothelial Injury in Sickle Cell Disease
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


