FIGURE 108-1. Genetic causes of mineralocorticoid deficiency. The schematic cell represents a renal cell from the distal tubule/collecting duct segment, which expresses the mineralocorticoid receptor (MR), is aldosterone (A) sensitive, and expresses 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), serum and glucocorticoid–regulated kinase 1 (sgk1), and the epithelial sodium channel (ENaC). CHIF, Channel-inducing factor; ERK, extracellular signal-regulated kinase; GILZ, glucocorticoid-induced leucine zipper protein.
Failure of Aldosterone Biosynthesis: Hypoaldosteronism
COMBINED GLUCOCORTICOID AND MINERALOCORTICOID DEFICIENCIES: ADRENAL FAILURE
The causes of primary adrenal failure and its clinical features are described elsewhere. Aldosterone deficiency occurs in congenital adrenal hyperplasia due to 21-hydroxylase and 3β-hydroxysteroid dehydrogenase deficiencies. Patients with 11β-hydroxylase or 17α-hydroxylase deficiency also have hypoaldosteronism but have mineralocorticoid excess because of excess secretion of the mineralocorticoid deoxycorticosterone (DOC) proximal to the enzymatic block. At presentation, most patients with primary autoimmune adrenal failure have evidence of both glucocorticoid and mineralocorticoid deficiency. However, as adrenal failure evolves, selective aldosterone deficiency can occur in the presence of preserved zona fasciculata function. While glucocorticoid responsiveness to ACTH, metyrapone, or insulin-induced hypoglycemia may be normal, plasma renin activity (PRA) is elevated, and plasma aldosterone levels are low or undetectable. This is accompanied by mild metabolic acidosis and, occasionally, hyponatremia. With time, progression to “panadrenal” insufficiency can occur. A year or more can separate the onset of the mineralocorticoid and glucocorticoid deficiencies.18–20
It is important to recognize that mineralocorticoid deficiency is not a feature of secondary adrenal failure, caused by hypothalamic or pituitary disease, because the renin-angiotensin system is not affected.
Primary adrenal hypoplasia often affects the zona glomerulosa, causing mineralocorticoid insufficiency and salt loss, as well as impaired glucocorticoid synthesis and release. Mutations in DAX1 (dosage-sensitive sex reversal adrenal hypoplasia congenita–critical region on the X chromosome gene 1) cause X-linked adrenal hypoplasia congenita (AHC).21 The patients are characterized by primary adrenal failure (combined glucocorticoid and mineralocorticoid deficiency), testicular dysgenesis, and gonadotropin deficiency. Most DAX1 mutations are deletions, nonsense, or frameshift mutations that markedly impair its transcriptional activity. Mild DAX1 mutations, such as the missense mutation (W105C) in the amino-terminal region of the DAX1 gene, are associated with more variable clinical phenotypes and may be a cause of isolated mineralocorticoid deficiency.22 Aldosterone deficiency also occurs in 10% to 15% of individuals who have ACTH resistance as part of the triple A (Allgrove) syndrome (AAAS, ALADIN), with isolated adrenal failure, alacrima, or upper-gastrointestinal abnormalities such as achalasia of the esophagus.23 Severe loss-of-function mutations in the MC2R (ACTH-receptor) gene, which is expressed also in the zona glomerulosa, may be found in a significant proportion of children with primary adrenal insufficiency, e.g., familial glucocorticoid deficiency type 1 (FGD1) and who have been diagnosed as having salt-losing forms of adrenal hypoplasia. These findings may suggest a supportive role for ACTH in mineralocorticoid synthesis and release, especially in times of stress (e.g., infection), salt-restriction, heat, or relative mineralocorticoid insensitivity.24 Mineralocorticoid requirements often decrease with age, as evidenced by the fall in normal mineralocorticoid secretion rates after infancy.
ISOLATED HYPOALDOSTERONISM
Selective deficiency of aldosterone secretion without alteration in cortisol production can result from inborn errors in aldosterone biosynthesis, failure of the zona-glomerulosa function of the adrenal gland secondary to drugs, surgery, or the syndrome of hyporeninemic hypoaldosteronism.
Primary Isolated Hypoaldosteronism
Prior to the characterization of the CYP11B2 gene, which is located on chromosome 8q24,25,26 the disease was termed corticosterone methyl oxidase type I (CMO I) deficiency and corticosterone methyl oxidase type II (CMO II) deficiency. Subsequently, both variants were shown to be secondary to mutations in aldosterone synthase and are now termed type 1 and type 2 aldosterone synthase deficiency. Aldosterone synthase catalyses the three terminal steps of aldosterone biosynthesis, 11β-hydroxylation of deoxycorticosterone to corticosterone, 18-hydroxylation to 18-hydroxycorticosterone, and 18-oxidation to aldosterone. Patients with type 1 aldosterone synthase deficiency have low to normal levels of 18-hydroxycorticosterone but undetectable levels of aldosterone (or urinary tetrahydroaldosterone), whereas patients with the type 2 variant have high levels of 18-hydroxycorticosterone and only subnormal or even normal levels of aldosterone. This suggests blockade of only the terminal 18-oxidation step, with some residual aldosterone synthase activity. The explanation for the variable biochemical phenotype is unknown, particularly now that the same mutation in the CYP11B2 gene has been uncovered in both variants (Fig. 108-2, Table 108-1). It is possible that this may reflect polymorphic variants in the residual and normal product of the CYP11B1 gene, 11β-hydroxylase.
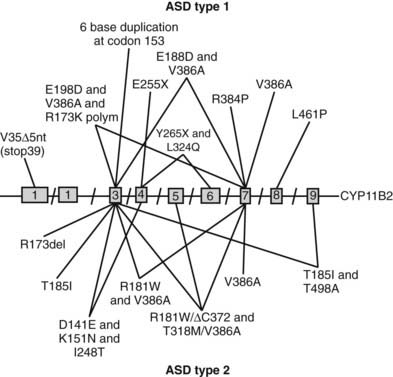
FIGURE 108-2. Schematic representation of identified mutations in the CYP11B2 gene. The CYP11B2 gene is represented with its intron/exon structure. Mutations found for aldosterone synthase deficiency (ASD) type 1 are shown above the gene structure; those responsible for ASD type 2 are presented below the gene structure.

At variance with the MR knockout mouse model, the aldosterone synthase knockout mouse model is not lethal. As expected, ionic homeostasis is altered in the absence of aldosterone, but high levels of corticosterone and angiotensin II seem to partially rescue sodium balance,27 underscoring the importance of the MR over its ligand, aldosterone.
Both variants of aldosterone synthase deficiency are rare and inherited as autosomal-recessive traits (see Table 108-1). The type 2 deficiency is found most frequently among Jews of Iranian origin.
The severity of the clinical manifestations is inversely related to the age at diagnosis in both variants. In most infants, the disorders become less severe as the child ages. Aldosterone synthase type 2 deficiency becomes clinically apparent between 1 week and 3 months of age, with severe dehydration, vomiting, and failure to grow and thrive. Hyponatremia, hyperkalemia, and metabolic acidosis are uniformly present. The plasma renin activity is elevated, and plasma aldosterone levels are low. Plasma 18-hydroxycorticosterone levels are markedly elevated, and the ratio of 18-hydroxycorticosterone to aldosterone in plasma exceeds 5. The ratio of the urinary metabolite of 18-hydroxycorticosterone, 18-OH-THA, to the urinary metabolite of aldosterone, TH-Aldo, is also greater than 5. In older children, adolescents, and adults, the abnormal steroid pattern described may be present and may persist throughout life without clinical manifestations. Recently, type 1 aldosterone synthase deficiency has been reported with onset in middle age. Mineralocorticoids are given (9α-fludrocortisone) during infancy and early childhood, but this therapy can be discontinued in the majority of adults. Spontaneous “normalization” of growth can occur in untreated patients. It is not understood why aldosterone deficiency is so much more threatening in infancy than in adult life. It is particularly puzzling that isolated hypoaldosteronism due to low renin secretion in aging patients has clinical significance, while patients with asymptomatic inherited hypoaldosteronism due to aldosterone synthase deficiency exhibit none of the manifestations of hyporeninemic hypoaldosteronism.
Secondary Isolated Hypoaldosteronism
Hyperreninemic hypoaldosteronism can occur in critically ill patients with disorders such as sepsis, cardiogenic shock, or liver cirrhosis.47,48 Cortisol levels in these patients are elevated and commensurate with the level of stress. In normal subjects, aldosterone, corticosterone, and 18-hydroxycorticosterone levels can be suppressed in 48 to 96 hours with continuous ACTH stimulation.49–51 Prolonged ACTH secretion is thought to impair aldosterone synthase activity and explain the underlying mechanism of this syndrome. These patients have an increased ratio of plasma 18-hydroxycorticosterone to aldosterone, and the aldosterone response to angiotensin II infusion is impaired. Hypoxia and proinflammatory cytokines may be additional mechanisms that inhibit aldosterone synthesis from the zona glomerulosa, as may elevated circulating levels of atrial natriuretic peptide (ANP).52 Additionally, many critically ill patients are taking medications that can interfere with aldosterone production (see later). Hyperreninemic hypoaldosteronism is also reported in patients with tumors that have metastasized to the adrenals.
Since no major clinical complications are reported with this form of hypoaldosteronism, it seldom requires treatment. Avoidance of drugs and factors that can exacerbate the condition, however, is essential.
SYNDROME OF HYPORENINEMIC HYPOALDOSTERONISM
The syndrome of hyporeninemic hypoaldosteronism (SHH), also referred to as type 4 distal renal tubular acidosis (RTA), is not uncommon and usually occurs in late middle age (median age 68 years) in males more than females. Underlying diabetes mellitus is present in 50% of cases and chronic renal insufficiency in 80% of cases. It is frequent in patients with tubulointerstitial forms of renal disease but has been described in virtually every type of renal abnormality.53–56 SHH accounts for 50% to 70% of patients with unexplained hyperkalemia and renal disease in patients with relatively preserved glomerular filtration rate (GFR).53,55,56
Patients have low PRA and aldosterone levels that cannot be stimulated by provocative tests. Hyperchloremic metabolic acidosis occurs in about 70%, and mild to moderate hyponatremia is seen in about 50% of affected patients. Hyperkalemia that is out of proportion to the degree of renal insufficiency is observed in all patients.55,56 The pathogenesis is unknown, but proposed mechanisms include hyporeninemia due to damaged juxtaglomerular apparatus, sympathetic insufficiency, altered renal prostaglandin production, and impaired conversion of prorenin to renin.57,58 Low renin production does not appear to be the sole factor, since PRA can be normal in some patients. It is possible that some cases can be explained by sodium retention leading to volume expansion, with secondary suppression of renin and aldosterone.
The leading causes of interstitial nephritis, in which hyperkalemia occurs early and before chronic renal failure, are anatomic genitourinary abnormalities, analgesic abuse with aspirin or phenacetin, hyperuricemia, nephrocalcinosis, nephrolithiasis, and sickle cell disease. Diabetic patients are predisposed to hyperkalemia because of insulin deficiency and hyperglycemia, and this may be exacerbated by autonomic neuropathy. IgM monoclonal gammopathy has been associated with nodular glomerulosclerosis, a concentrating defect, and hyporeninemic hypoaldosteronism.59 Patients with acquired immunodeficiency syndrome (AIDS) can have persistent hyperkalemia secondary to adrenal insufficiency or, less frequently, hyporeninemic hypoaldosteronism.
There is no ideal medical therapy for SHH. The majority of patients with mild selective hypoaldosteronism require no therapy. Treatment may be indicated in patients with severe hyperkalemia. Reducing the extracellular potassium load is the single most effective measure in controlling hyperkalemia. Reducing dietary intake of potassium is helpful. The long-term control of glucose homeostasis in diabetes mellitus may reduce the risk of developing SHH, and autonomic insufficiency is probably avoidable in well-controlled diabetes. Since many medications can interfere with the renin-aldosterone axis, avoidance of these drugs is of major importance. β-Adrenergic receptor blockers, prostaglandin synthetase inhibitors, and potassium-sparing diuretics should be avoided in patients with SHH and in diabetic patients with latent hypoaldosteronism. Calcium channel blockers, antidopaminergic agents, and drugs that impair adrenal function must be used with caution. Patients taking angiotensin-converting enzyme (ACE) inhibitors must be monitored carefully for hyperkalemia. The prolonged administration of heparin can worsen hypoaldosteronism60 and has been associated with lethal hyperkalemia. Fludrocortisone 0.2 mg/day for 2 weeks usually normalizes potassium levels in patients with SHH,61,62 but there is a risk of salt retention and hypertension. In severe SHH, fludrocortisone acetate, in doses of 0.1 to 1.0 mg/day (equivalent to 200 to 2000 µg aldosterone daily), can be required. Diuretics may be the optimal therapy for patients with SHH and coexisting diseases associated with sodium retention. Older patients with hypertension, mild renal impairment, and congestive heart failure respond better to diuretic therapy than to mineralocorticoid replacement. Since kaliuresis is the goal of diuretic therapy, the diuretic employed should have a potent kaliuretic activity; thiazide diuretics are more effective than loop diuretics and induce less natriuresis.
POSTADRENALECTOMY HYPOALDOSTERONISM
In a patient with a unilateral aldosteronoma (Conn’s syndrome), the contralateral zona glomerulosa is frequently suppressed. Without reversal of the chronic volume expansion preoperatively, postadrenalectomy patients may develop severe hyperkalemia and hypotension lasting several days to several weeks after surgery. This may be exacerbated by the use of spironolactone preoperatively. This drug has a long half-life and should be discontinued 2 to 3 days before surgery to minimize the risk of mineralocorticoid deficiency postoperatively.
PHARMACOLOGIC INHIBITION OF ALDOSTERONE
Cyclosporin, heparin sodium, and calcium channel blockers specifically inhibit aldosterone production from the zona glomerulosa. Cyclosporin blocks angiotensin II–induced aldosterone production and inhibits “growth” and steroidogenic capacity of adrenocortical cells.63 The latter effect may be caused by an impairment of protein synthesis. Additionally, cyclosporin and tacrolimus (FK506) inhibit MR transcription activity without affecting aldosterone binding.64 Polysulfated glycosaminoglycans, such as heparin sodium, impair aldosterone biosynthesis. With prolonged administration, heparin sodium can produce significant hypoaldosteronism with severe hyperkalemia65 because of a direct toxic effect on the zona glomerulosa, evidenced by a hyperreninemic hypoaldosteronism and zona glomerulosa atrophy. The least toxic dose of heparin sodium is unknown, but doses as small as 20,000 units/day for 5 days can reduce aldosterone secretion. This is an uncommon cause of hypoaldosteronism but is important to recognize because it is reversible and can be lethal. The offending agent seems to be chlorobutanol, used as a preservative for heparin, rather than the heparin molecule itself. Calcium channel blockers inhibit aldosterone production and under certain clinical conditions can impair aldosterone secretion by inhibiting calcium influx. β-Blockers and prostaglandin synthase inhibitors are frequent causes of hyporeninemic hypoaldosteronism. β-Blockers inhibit renin secretion from the juxtaglomerular apparatus, and prostaglandin synthetase inhibitors, which specifically inhibit cyclooxygenase, block renin release. ACE inhibitors and potassium-sparing diuretics can contribute to hypoaldosteronism and resultant hyperkalemia. ACE inhibitors “inhibit” ACE, thus interrupting the renin-aldosterone axis and leading to iatrogenic hypoaldosteronism. Spironolactone has two effects: it is a mineralocorticoid-receptor antagonist, and it inhibits aldosterone biosynthesis. Triamterene causes potassium retention by a direct action on non-aldosterone-mediated, distal-tubular exchange sites. Amiloride acts on the luminal surfaces of epithelial membranes to block sodium channels, resulting in less sodium resorption and potassium secretion. Drugs that impair adrenal function are increasingly used for the hormonal treatment of breast cancer and medical management of Cushing’s syndrome. Most can cause hypoaldosteronism. Aminoglutethimide, metyrapone, and trilostane block various enzymatic steps in the synthesis of mineralocorticoids, glucocorticoids, and adrenal sex steroids. Lower doses of these drugs may not be associated with hyperkalemia, since aldosterone precursors such as deoxycorticosterone may supply the necessary mineralocorticoid activity. Finally, drugs affecting the dopaminergic system can produce significant alterations in aldosterone secretion. It is believed that aldosterone is under tonic dopamine inhibition. Thus, dopaminergic agonists such as bromocriptine may impair aldosterone secretion in certain physiologic situations.
Failure of Aldosterone Action: Mineralocorticoid Resistance
Mineralocorticoid resistance implies a lack of response to aldosterone despite normal or even increased concentrations. The classification of this entity has been confusing, but the elucidation of the molecular basis of many of these conditions has clarified the field. Two main types of so-called pseudohypoaldosteronism fulfill the criteria for mineralocorticoid resistance.
PSEUDOHYPOALDOSTERONISM
Pseudohypoaldosteronism (PHA) is a rare, inherited salt-wasting disorder first described by Cheek and Perry in 1958 as a defective renal tubular response to mineralocorticoid in infancy. Patients present in the neonatal period with dehydration, hyponatremia, hyperkalemia, metabolic acidosis, and failure to thrive despite normal glomerular filtration and normal renal and adrenal function.66 Renin levels and plasma aldosterone are grossly elevated. When patients fail to respond to mineralocorticoid therapy, PHA is suspected as the underlying disorder.
PHA type 1 can be divided into two distinct disorders based on unique physiologic and genetic characteristics: the renal form of PHA, inherited as an autosomal dominant (AD) trait, and a generalized autosomal recessive (AR) form of PHA. Some de novo cases are described as sporadic. The AD form is usually less severe, with the patient’s condition often improving spontaneously within the first several years of life, thus allowing discontinuation of therapy and treatment. Adult patients with the AD form are clinically indistinguishable from their wild-type relatives except for presumably lifelong elevation of renin, angiotensin II, and aldosterone levels. However, it has been suggested that the seemingly benign AD form may have been a fatal neonatal disorder in previous eras, preventing propagation of disease alleles.67 By contrast, the AR form has a multiorgan disorder, with mineralocorticoid resistance seen in the kidney, sweat and salivary glands, and the colonic mucosa.68 The latter condition does not spontaneously improve with age.69 As a result, this form is considered to be more severe because it usually persists into adulthood. Since sodium reabsorption is coupled to potassium and hydrogen ion secretion, patients often show decreased potassium and hydrogen ion secretion with decreased sodium reabsorption. Hence, potassium and hydrogen ions accumulate in the body, and this ultimately leads to hyperkalemia and metabolic acidosis. Moreover, a decrease in vascular volume leads to activation of the renin-angiotensin-aldosterone axis.
The underlying basis for the AD form of PHA is explained on the basis of different heterozygous inactivating mutations in the mineralocorticoid receptor (MR) (Table 108-2). One mutated allele of the MR gene is sufficient to lead to the renal phenotype in men. In contrast, MR knockout mice showed hyponatremia, hyperkalemia, a strongly activated renin-angiotensin-aldosterone system (RAAS), significantly reduced ENaC activity in kidney and colon, and the mice died between day 8 and day 13 after birth when they were not treated with isotonic NaCl solutions.70 By contrast, heterozygous MR knockout mice grow and breed normally and show no salt wasting. This difference between humans and mice might be due to differences in neonatal kidney maturation. However, only heterozygous mutations have been reported in humans, suggesting that the homozygous state may be embryologically lethal. The loss of one allele results in haploinsufficiency sufficient to generate the AD form of PHA symptoms, thus underlining the importance of a substantial MR protein level, most notably during the neonatal period.67
Table 108-2. Mineralocorticoid Receptor Mutations in Patients With the Autosomal-Dominant Form of Pseudohypoaldosteronism
| Mutation | Location | Author |
|---|---|---|
| c.215G>C (−2 in Kozak seq. of translation initiation site) and c.754A>G (Ile180Val) and c.938C>T (Ala241Val) | Intron 1 Exon 2 Exon 2 | 72 |
| c.402T>A (Y134X stop) nonsense | Exon 2 | 75 |
| c.488C>G (S163stop) nonsense | Exon 2 | 76 |
| del8bp537; frameshift | Exon 2 | 77 |
| c.981delC (pSer328 frameshift) | Exon 2 | 74 |
| c.1029C>A (Tyr343stop); nonsense | Exon 2 | 74, 78 |
| c.1131dupT (E378X stop) | Exon 2 | 79 |
| ΔG1226; frameshift leads to premature stop codon | Exon 2 | 7 |
| c.1308T>A (C436stop); nonsense | Exon 2 | 80 |
| InsT1354; frameshift | Exon 2 | 77 |
| ΔT1597; frameshift leads to premature stop codon | Exon 2 | 71 |
| InsA1715 (Y503Xstop); heterozygous | Exon 2 | 67 |
| c.1831C>T (R537stop); nonsense | Exon 2 | 71 |
| c.1679G>A (pTrp560Xstop) | Exon 2 | 74 |
| c.1757+1G>A; splice donor site | Intron B | 78 |
| c.1984C>T (Arg590Xstop); heterozygous | Exon 3 | 67 |
| c.1768C>T (pArg590Xstop) | Exon 3 | 74 |
| c.1811delT (pLeu604 frameshift) | Exon 3 | 74 |
| c.2119G>A (G633R); missense | Exon 3 | 77 |
| c.2157C>A (Cys645stop); nonsense | Exon 4 | 77 |
| c.1934G>C (pCys645Ser) | Exon 4 | 74 |
| c.1954C>T (Arg652stop); nonsense | Exon 4 | 74, 78 |
| c.1977A>C (pArg659Ser) | Exon 4 | 74 |
| c.2017C>T (R673Xstop) | Exon 5 | 79 |
| c.2020A>T (pLys674Xstop) | Exon 5 | 74 |
| c.2024C>g (S675Xstop) | Exon 5 | 79 |
| c.2125delA, frameshift T709 leads to L772Xstop | Exon 5 | 75 |
| c.2275C>T (pPro759Ser) | Exon 5 | 74 |
| c.2306_2307inv (pLeu769Pro) | Exon 5 | 74 |
| c.2310C>A (p.Asn770Lys); missense | Exon 5 | 74, 78 |
| c.2549A>G (Q776R); missense | Exon 5 | 77 |
| c.2581G>A; splice alteration – nonsense, heterozygous | Exon 5 | 79 |
| ΔA; aberrant splicing | Intron 5 | 71 |
| c.2413T>C (pSer805Pro) | Exon 6 | 74 |
| c.2445C>A (pSer815Arg) | Exon 6 | 74 |
| c.2669C>T or c2453C>T (S818L); missense, heterozygous | Exon 6 | 67, 79 |
| InsA2681 (fsH821); frameshift, heterozygous | Exon 6 | 67 |
| c.2771T>C (L924P); nonsense | Exon 8 | 81 |
| c.2779+1G>A; abnormal splicing | Exon 8 | 74 |
| c.2839C>T or c.3055C>T (R947Xstop) | Exon 9 | 73, 82 |
| InsC2871; frameshift from codon 958 | Exon 9 | 79, 83 |
| c.3115C>T (Q967stop); heterozygous | Exon 9 | 67 |
| c.2915A>G (E972G) | Exon 9 | 79 |
| c.3158T>C (L979P); missense | Exon 9 | 77 |
To date, more than 40 mutations in the MR gene have been described in patients with the AD form of PHA (see Table 108-2; Fig. 108-3). They include missense, nonsense, frameshift, and splice-site mutations, as well as deletions spread throughout the gene. These mutations are responsible for either an early termination of translation with MR truncation or a defect in MR activity (loss of LBD or DBD), disruption of nucleocytoplasmic shuttling, or alteration in some transcriptional coregulator recruitment. The clinical improvement after infancy cannot yet be explained on the molecular level. The hypothesis that polymorphisms within the three ENaC gene subunits are responsible could not be shown, but today potential candidates might be the ubiquitin protein ligase Nedd4 and the serine-threonine kinases WNK1 and WNK4.
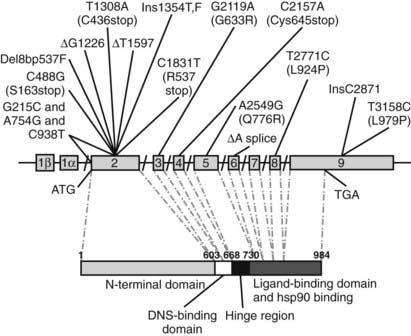
FIGURE 108-3. Schematic representation of identified mutations in the mineralocorticoid receptor (MR) gene. The MR gene is represented with its intron/exon structure. Eight exons (2 to 9) code for the functional domains, which are shown below in the amino acid sequence. The translation initiation site (ATG) and the translation stop codon (TGA) are shown.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


