Ligand
Functions
Copies/platelet
Integrins
αIIbβ3 (GPIIb-IIIa)
Fibrinogen
Aggregation, adhesion
~80,000
α2β1 (GPIa-IIa)
Collagen
Adhesion
3000–5000
α5β1 (VLA-5)
Fibronectin
Adhesion
1000
α6β1 (VLA-6)
Laminin
Adhesion
1000
αvβ3
Vitronectin
Adhesion
100
Leucine-rich glycoproteins
GPIb-IX
von Willebrand factor
Adhesion
25,000
GPV
12,500
Immunoglobulin cell adhesion molecules
GPVI
Collagen
Adhesion, activation
4000–6000
PECAM-1 (CD31)
PECAM-1
Adhesion
8000
Fcg-RII (CD32)
Immune complex binding
~1000
Selectins
P-selectin (CD62P)
PSGL-1
Platelet-leukocyte adhesion
20,000
Miscellaneous
GPIV (CD36)
Collagen, thrombospondin, oxidized LDL
20,000
CLEC-2
Podoplanin
Adhesion/activation
At the site of blood vessel injury, platelet glycoproteins mediate a series of platelet reactions: rolling, tethering, adhesion, and aggregation (Fig. 1). First, circulating platelets form indirect interaction with vessel wall through von Willebrand factor (VWF). Plasma VWF bound to exposed collagen forms a transient bridge between collagen and platelet GPIb. During platelet rolling and tethering on collagen, additional direct collagen-platelet interactions are induced via platelet-collagen receptors GPIa-IIa (integrin α2β1) and GPVI for firm adhesion. Collagen-bound GPVI initiates intracellular signaling leading to cytoskeletal rearrangement, calcium mobilization, and granule release. In addition to ADP and thromboxane A2 (TXA2) released from activated platelets, thrombin generated by the coagulation cascade stimulates platelets as a secondary agonist for further activation. These secondary agonists act through G protein-coupled receptors and induce “inside-out signaling” for fibrinogen receptor GPIIb-IIIa (integrin αIIbβ3) activation which cross-links platelets and leads to platelet aggregation (primary hemostasis).
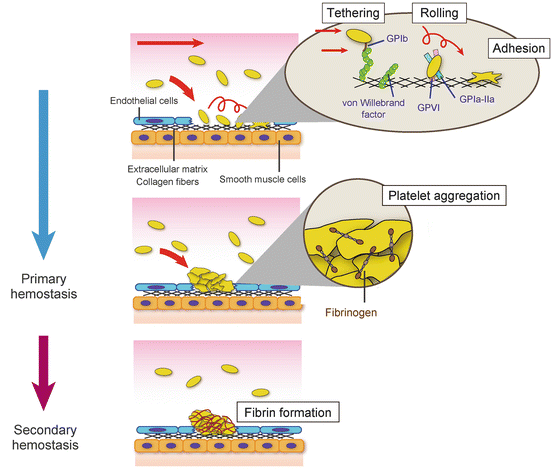
Fig. 1
Primary and secondary hemostasis. At the site of vascular injury, von Willebrand factor mediates transient and indirect interaction of circulating platelets with exposed subendothelial collagen. During platelet rolling on collagen, platelet-collagen receptors, GPVI and GPIa-IIa, form stable direct interaction with collagen followed by firm platelet adhesion. Platelets are activated by thrombin, ADP, and thromboxane A2, and activated GPIIb-IIIa (integrin αIIbβ3) by inside-out signaling forms aggregation via fibrinogen binding (primary hemostasis). In secondary hemostasis, fibrin which is produced by coagulation cascade stabilizes the thrombus formed in primary hemostasis
Following platelet aggregation, insoluble fibrin, which is generated by the coagulation cascade, incorporates into platelet aggregation and stabilizes the platelet aggregation (secondary hemostasis).
2 GPIIb-IIIa (Integrin αIIbβ3)
The fibrinogen receptor GPIIb-IIIa is the major platelet surface glycoprotein which expression is restricted to platelets and megakaryocytes [1]. Each platelet expresses about 80,000 copies of GPIIb-IIIa on their surface [2], and the indispensable role of GPIIb-IIIa for platelet aggregation and hemostasis has been established from the studies of Glanzmann thrombasthenia (GT). GT was first reported in 1918 from a series of patients with an inherited bleeding disorder, and polyacrylamide gel electrophoresis revealed that GPIIb and GPIIIa are deficient in platelets of GT [3, 4]. In 1986, Tamkun et al. started using the word “integrin” for cell surface transmembrane receptors which mediate the linkage between extracellular matrix and cytoskeleton [5]. Integrins are heterodimers of non-covalently associated α- and β-subunits. Twenty-four different integrins are formed from 18 α- and 8 β-subunits, and now GPIIb-IIIa is designated as “integrin αIIbβ3.”
2.1 Integrin αIIbβ3 Structure
αIIbβ3 is a typical integrin consisting of αIIb and β3 heterodimer (Fig. 2a). αIIb is a 140 kDa glycoprotein [6] which is composed of a disulfide bond connected to a heavy chain (114 kDa, 871 amino acids) and a light chain (23 kDa, 137 amino acids) [7]. β3 is a 116 kDa glycoprotein [8] (762 amino acids), which is non-covalently linked to αIIb. In megakaryocytes, αIIb is synthesized as a single chain (pro-αIIb) and associates with β3 in the endoplasmic reticulum. The pro-αIIb/β3 complex is transported to the Golgi apparatus, and the proteolytic cleavage of pro-αIIb/β3 yields the mature αIIbβ3. The extracellular domain of αIIb is composed of a seven-bladed β-propeller domain containing ligand-binding sites, thigh domain, calf-1 domain, and calf-2 domain. β3 is composed of βA domain containing ligand-binding sites, plexin/semaphorin/integrin (PSI) domain, hybrid domain, four epidermal growth factor (EGF) repeats, and membrane proximal β-tail domain (βTD) (Fig. 2b). By stable non-covalent association, αIIbβ3 forms an 8 × 12 nm globular head which contains ligand-binding site with 18 nm two rodlike flexible tails [9]. Following the studies of αvβ3 [10, 11], X-ray crystallography revealed the bent conformation of αIIbβ3 [12]. This bent conformation is achieved by the flexibility at the “genu” located between the thigh and calf-1 domain in αIIb and EGF1 and EGF2 in β3. In bent conformation, the ligand-binding face at the globular head is not exposed, and the natural ligands, such as fibrinogen, are not allowed to interact with αIIbβ3. Upon activation, αIIbβ3 can rapidly change its conformation from bent to extended form which is high affinity for fibrinogen (Fig. 2c). Following the short single-spanning transmembrane region, both αIIb- and β3-subunits have short cytoplasmic tail which is important for conformational change of αIIbβ3 (Fig. 2d). In the membrane proximal region, arginine (R) and aspartic acid (D) in the conserved αIIb-GFFKR and β3-HDRKE sequence form a salt bridge. This salt bridge maintains αIIbβ3 in low-affinity inactive state, and αIIbβ3 becomes constitutively active by disruption of the salt bridge [13–16]. Even integrin cytoplasmic domain has no enzymatic or actin-binding activity, and the molecule interactions with cytoplasmic region induce signals for platelet activation [17, 18]. Especially two well-conserved NPXY motifs in β3 tail, NPLY (744–747) and NITY (756–759), are essential for talin and kindling-3 binding, respectively [19]. The last three amino acid residues of β3 is the important binding site for c-Src [20–23].
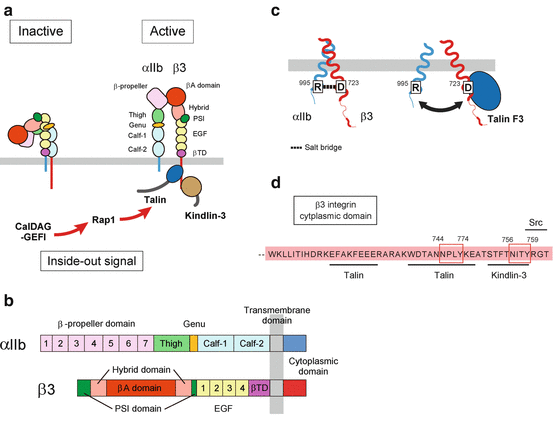
Fig. 2
The structure and inside-out activation of integrin αIIβ3. (a, b) The domain structure and conformational change of integrin αIIβ3. When αIIbβ3 is in a low-affinity inactive state, αIIbβ3 extracellular domain is in bent conformation. However, once αIIbβ3 is activated by inside-out signaling, talin and kindling-3 interaction with β3 cytoplasmic tail induces conformational change of extracellular domain of αIIbβ3 resulting in extended conformation. (c) Disruption of salt bridge in cytoplasmic domain of αIIbβ3 by talin binding. In inactive state αIIbβ3, the salt bridge formed between R995 of αIIb and D723 of β3 is important to keep αIIbβ3 inactive. Inside-out signaling which induced binding of talin head domain to β3 cytoplasmic tail disrupts the salt bridge and induces conformational change of αIIbβ3. (d) The binding site of talin, kindlin-3, and Src in β3 cytoplasmic domain
2.2 αIIbβ3 Inside-Out Signaling
The affinity and conformation of αIIbβ3 are tightly regulated by inside-out signaling [24, 25]. Although the precise molecular mechanism of inside-out signaling remains unknown, the direct interaction of talin with β3 cytoplasmic tail is essential at the final step of inside-out αIIbβ3 activation [26–30]. Talin is a 250 kDa actin-binding protein which is composed of a 50 kDa head domain and a 220 kDa rod domain. The N-terminal head domain contains a FERM (band four-point one, ezrin, radixin, and moesin) domain which is further divided into F1, F2, and F3 subdomains [31]. Talin F3 interaction with β3 cytoplasmic first NPXY motif (744–747) [28] disrupts the αIIb-R995/β3-D723 slat bridge followed by the conformational change for αIIbβ3 activation. In genetically modified mice with impaired talin/β3 interaction or talin deficiency, agonist-induced inside-out αIIbβ3 activation is strongly impaired resulting in bleeding diathesis [32–35]. Another FERM domain containing protein kindlin-3 is also essential for αIIbβ3 activation, and deficiency of kindlin-3 is associated with bleeding tendency [36, 37]. In contrast to talin, kindlin-3 F3 domain binds to the membrane-distal second NPXY motif (756–759) [38–40]. Although both talin and kindlin-3 are required for inside-out αIIbβ3 activation, kindlin-3 alone is insufficient for αIIbβ3 activation [41, 42], and probably kindlin-3 cooperates with talin by increasing the avidity of αIIbβ3 [43, 44]. Although no patient with mutation in talin has been reported, mutation in kindling-3 is known to be a cause of leukocyte adhesion deficiency (LAD)-III [45]. In LAD-III, impaired integrin activation in platelets and leukocytes causes bleeding problem and immune defect.
A Ras family member small GTPase Rap1 [46, 47] and calcium- and diacylglycerol-regulated guanine exchange factor-1 (CalDAG-GEFI) [48–51] are the critical molecules at the upstream signaling of talin/β3 interaction. CalDAG-GEFI activates Rap1 by facilitating the release of bound GDP, and activated GTP-bound Rap1 recruits talin to β3 cytoplasmic tail followed by αIIbβ3 activation [52]. Recently, CalDAG-GEFI-deficient patients [53, 54] or a patient expressing CalDAG-GEFI with loss-of-function mutation which causes severe bleeding problems was reported [55].
2.3 Clinical Importance of αIIbβ3
GT is an autosomal recessive inherited bleeding disorder due to a quantitative and a qualitative defect in αIIbβ3 [56]. The impaired fibrinogen binding to αIIbβ3 leads to bleeding symptoms such as purpura, petechiae, easy bruising, mucocutaneous bleeding, and excessive bleeding after injury or surgical procedure [57]. In platelet aggregation assay, aggregation response is absent in response to ADP, thrombin, collagen, or arachidonic acid in platelets of patient with typical GT [58]. However, ristocetin-induced aggregation response is induced normally. Flow cytometric analysis of agonist-induced αIIbβ3 activation by binding of activated αIIbβ3-specific monoclonal antibody PAC-1 [59] and molecular genetic analysis [60, 61] of αIIb and β3 is the key to diagnosis. Based on the expression levels of αIIbβ3, GT is classified into three types. The most severe type I platelets express <5%, and type II express 5–20% αIIbβ3 of normal levels. In type III, variant type, platelets express >20% of αIIbβ3. However, expressed αIIbβ3 is functionally impaired and does not support platelet aggregation. Currently, besides platelet transfusion, there is no specific treatment for GT, and the bleeding in GT patients is managed by antifibrinolytic therapy and platelet transfusion, and recombinant activated factor VII is performed [57, 62–64]. In future therapeutic strategy, gene therapy is expected to be a curative treatment for patients with GT [65–67].
3 GPIb-IX-V
Glycoprotein GPIb-IX-V complex is exclusively expressed on platelets and megakaryocytes and an essential receptor for the first step of hemostasis. This essential role of GPIb-IX-V is exemplified by the rare inherited bleeding disorder Bernard-Soulier syndrome (BSS) which was first reported and described by Jean Bernard and Jean Pierre Soulier in 1948. The acrylamide gel electrophoresis clearly showed the absence of GPIb band in platelets of BSS patient in the 1970s [68]. The clarification that additional two glycoproteins GPV and GPIX are missing in BSS platelets [69] and the cloning of GPIbα [70], GPIbβ [71], GPIX [72], and GPV [11, 12] achieved further develops on the role of GPIb-IX-V.
3.1 GPIb-IX-V Structure
GPIb-IX-V complex (Fig. 3) is the second most abundant platelet glycoprotein (25,000 copies per platelet [73]) next to fibrinogen receptor GPIIb-IIIa. GPIb-IX-V complex consists of four different type I transmembrane subunits, GPIbα (CD42b; 135kDa, 610 amino acids), GPIbβ (CD42c; 26kDa, 181 amino acids), GPIX (20 kDa, 160 amino acids), and GPV (82 kDa, 560 amino acids), with a stoichiometry of 2:2:2:1. Each subunit belongs to the leucine-rich repeat (LRR) protein family which is known to be involved in protein-protein interactions. LRR is the protein structure motif composed of 20–30 amino acid sequences, and the tandem repeat of LRR motifs folds into a horseshoe shape. GPIbα is the largest subunit of the complex, and the N-terminal membrane-distal 282 residues which are composed of seven LRRs are the binding site for VWF. VWF binding region is followed by acidic residue-rich sequence containing sulfated tyrosine, a heavily O-glycosylated macroglycopeptide domain, and stalk region. The extracellular domain is connected to a single transmembrane region and cytoplasmic tail. Both GPIbβ and GPIX are relatively smaller subunits which have a single LRR in the extracellular domain. In contrast to the high sequence similarity of extracellular domain between GPIbβ and GPIX, the short GPIX cytoplasmic tail consisting of five amino acids is different from that of GPIbβ (34 amino acids).
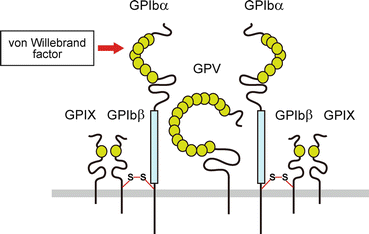
Fig. 3
Schematic representation of the GPIb-IX-V complex. GPIbα, GPIbβ, GPIX, and GPV form GPIb-IX-V complex at a stoichiometry of 2:2:2:1. The N-terminal of GPIbα contains binding sites for von Willebrand factor
3.2 GPIb-IX-V Function
The primary function of GPIb-IX-V is mediating initial platelet attachment to the blood vessel wall by interacting with VWF bound on exposed subendothelial collagen. The synthesized VWF in endothelial cells is stored in Weibel-Palade bodies or secreted into plasma. VWF circulates in plasma as multimer in various sizes ranging from 500 kDa to over 10,000 kDa. The platelet-binding potential of VWF with GPIbα is dependent on the multimer size and conformation of VWF which are regulated by metalloproteinase ADAMTS13 [74] and fluid shear stress [75], respectively. The deficient ADAMTS13 activity results in circulating the unusually large VWF multimers and causes thrombotic thrombocytopenic purpura (TTP) [76, 77]. Although the signaling pathway mediated by GPIb-IX-V for platelet activation remains unclear, VWF/GPIb-IX-V mediated a signaling pathway for platelet activation in which Src family kinase and glycolipid-enriched microdomains (GEMs) are involved [78].
3.3 Clinical Importance of GPIb-IX-V
Bernard-Soulier syndrome (BSS) is a rare autosomal recessive disease due to the genetic defects in GPIBA, GPIBB, and GP9 [79, 80] and characterized by bleeding tendency, giant platelets, and low platelet counts (macrothrombocytopenia) [81]. Due to the impaired VWF-mediated initial platelet interaction with exposed collagen, patients show variety of bleeding symptoms from early childhood. Although most mutations in BSS are associated with decreased surface expression or loss of function of GPIb-IX-V, platelet-type von Willebrand disease (VWD), an autosomal dominant disorder, results in gain-of-function phenotype [82–84]. In patients with platelet-type VWD, an excessive spontaneous VWF/GPIb interaction which accelerates the clearance of plasma VWF leads to the bleeding tendency. For the diagnosis of BSS, an isolated defect in ristocetin-induced platelet aggregation is the characteristic abnormality. Flow cytometric analysis of the surface expression levels of GPIb-IX-V, platelet counts, and examination of blood smear to determine large platelets are also important to confirm diagnosis.
4 Collagen Receptors Present on Platelets
Collagen is the major subendothelial protein which is essential for platelet adhesion and aggregation at the site of vascular injury. Platelets express two collagen receptors, GPIa-IIa (integrin α2β1) and GPVI. Although the roles of each collagen receptor in platelet functions have been extensively studied, the respective roles of GPVI and GPIa-IIa are still not clear. However, GPVI is believed as a central platelet-collagen receptor [85, 86], and GPIa-IIa is considered as a secondary collagen receptor to support platelet firm adhesion on collagen.
4.1 Introduction of GPVI
GPVI was first identified as a 62 kDa platelet protein recognized by autoantibody from the patient with idiopathic thrombocytopenic purpura (ITP) suffering from recurrent bleeding problem regardless of normal platelet count with corticosteroid treatment [87]. The patient’s platelets exhibited defective aggregation to collagen, and the 62 kDa protein was later identified as GPVI [88]. GPVI expression is restricted to platelets and megakaryocytes [89, 90] with varying GPVI expression levels [91–93].
4.2 GPVI Structure and Signaling Pathway
GPVI was cloned in 2000 and identified as a type I transmembrane glycoprotein which belongs to the immunoglobulin (Ig) superfamily [89] (Fig. 4). GPVI (62 kDa, 319 amino acids) is expressed at 4000–6000 copies per platelets as a complex with FcRγ subunit [94, 95]. GPVI has two Ig-like domains (D1 and D2) and a highly O-glycosylated mucin-rich stalk region in its extracellular domain. A 19-amino acid transmembrane domain is critical for linking to FcRγ subunit by salt bridge between GPVI Arg272 and Asp11 of FcRγ. A 51-amino acid cytoplasmic tail contains a calmodulin-binding basic motif, and constitutive calmodulin binding protects GPVI from shedding in resting platelets [96]. Upon platelet activation, calmodulin dissociation from GPVI leads to the cleavage of GPVI by metalloproteinase which results in decrease of GPVI expression probably to control platelet reactivity. The circulating 55 kDa GPVI ectodomain fragment could be a marker for platelet activation state. Proline-rich domain in cytoplasmic domain provides binding site for Src family kinase, Fyn and Lyn, which are critical for downstream signaling events through FcRγ chain [23]. Following the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) in FcRγ by Fyn and Lyn, the recruited tyrosine kinase Syk forms a signaling complex with adaptor proteins (SLP-67 and LAT) and phospholipase Cγ2 (PLCγ2) which leads to calcium mobilization, integrin activation, and granule release for further platelet activation [78, 97, 98].
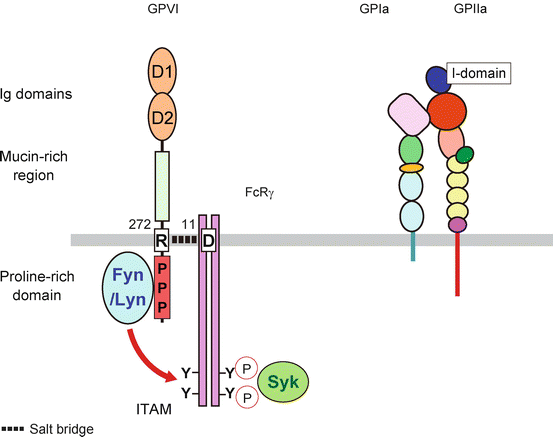
Fig. 4
Collagen receptor GPVI and GPIa-IIa. GPVI consists of two Ig domains followed by mucin-rich region in its extracellular domain. The R272 in transmembrane domain is important to form salt bridge with FcRγ chain. The binding of Src family kinase Fyn and Lyn phosphorylates the tyrosine in ITAM in cytoplasmic domain of FcRγ chain which leads to the Syk interaction and downstream signaling cascade. The extracellular domain of GPIIa has I-domain which is important for ligand binding
4.3 Clinical Importance of GPVI
GPVI deficiency is caused by congenital mutation [99–102] and acquired defect by immunodepletion with anti-GPVI autoantibody [87, 103–105] which is more frequent compared to congenital GPVI deficiency. Platelets from GPVI-deficient patients are defective for collagen-induced platelet aggregation in vitro. Although GPVI is recognized as a central receptor for firm platelet adhesion to collagen, human patients with GPVI deficiency usually have only mild bleeding problems, and their bleeding time is almost normal. Similar to human patients, GPVI-deficient mice generated by genetic knockout of GP6 gene [106] or immunodepletion by anti-GPVI antibody JAQ1 [107] also show only minor impact on bleeding time, suggesting that GPVI is not essential for normal hemostasis. However, in the lethal thromboembolism model by intravenous injection of collagen and epinephrine, significantly higher survival of GPVI-deficient mice [107, 108] indicates the crucial role of GPVI in arterial thrombosis. In fact, platelet adhesion and thrombus formation were reduced in GPVI-deficient mice after arterial injury induced by laser [109], FeCl3 [109, 110], wire, and vessel ligation [111]. In addition, GPVI polymorphisms in healthy subjects [112] are associated with stroke and cardiovascular events [113–117], and now the protective effect of GPVI deficiency in arterial thrombosis, not in normal hemostasis, suggests that GPVI may be an attractive target for antithrombotic therapies [118, 119].
4.4 GPIa-IIa (Integrin α2β1)
GPIa-IIa, which is now also designated as very late antigen-2 (VLA-2) [120] or integrin α2β1, was first recognized as a platelet-collagen receptor from the patient with bleeding problem in 1985 [121]. Following the cloning of GPIa [122] and GPIIa [123], the studies revealed the supportive role of GPIa-IIa in platelet adhesion on collagen.
GPIa-IIa, integrin α2β1, is a member of integrin family and consists of non-covalently associated α2 and β1 integrin, and platelets express 3000–5000 copies of GPIa-IIa per platelet. The extracellular domain of α2 integrin has 220 amino acid insertions, called I-domain, in the N-terminal β-propeller region. The sequence of I-domain is homologous to the collagen binding region of other proteins such as VWF and critical for α2β1 interaction with collagen types I, II, IV, and XI [124, 125].
4.5 GPIa-IIa Function and Clinical Importance
In patients with decreased GPIa, platelets are not responsive to collagen stimulation in aggregation study [121]. The affinity of GPIa-IIa for collagen is regulated by inside-out signaling, and agonist stimulation increases the binding of collagen to GPIa-IIa [126–128]. In contrast to GPIa-IIa, another platelet-collagen receptor GPVI does not need to be pre-activated for collagen interaction, and GPVI-collagen interaction induces intracellular signals for GPIa-IIa activation [86]. The number of GPIa-IIa on platelets is related to platelet response to collagen in normal population [129]. As shown in GPIa polymorphism C807T [130], GPIa polymorphism is associated with the expression levels of GPIa-IIa, and the linkage between GPIa-IIa expression levels [115] and polymorphism with thrombotic event [131, 132] is reported, suggesting the importance of GPIa-IIa in clinical outcomes. In experimental thrombus formation in vivo, the thrombus formed in the carotid artery by laser injury was significantly decreased in GPIa-IIa-deficient mice [133, 134].
5 Conclusions
Platelets are involved in both normal hemostasis and pathological thrombosis such as myocardial infarction and ischemic stroke. Platelet membrane glycoproteins are essential as adhesion molecules for platelet functions to communicate with the blood vessel wall and other cells. Starting from the analysis of patients with bleeding symptoms, the technical development and the application of genetically modified mice revealed the role of membrane glycoproteins in the process of thrombus formation. Further elucidation of the detail of platelet functions is expected from the recent genomic and proteomic techniques.
References
1.
Coller BS, Shattil SJ. The GPIIb/IIIa (integrin alphaIIbbeta3) odyssey: a technology-driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112:3011–25.PubMedPubMedCentral
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


