Fig. 1
Comparison between platelet-associated IgG (PAIgG) and autoantigen-specific antibody in ITP. In addition to autoantibody, PAIgG could contain an immune complex bound to the platelet Fcγ receptor (FcγRII) and nonspecific IgG bound to platelets, which results in the low specificity of PAIgG for the diagnosis of ITP
To overcome the disadvantage of PAIgG measurement and reveal the pathophysiology of ITP, an autoantigen that binds autoantibody should be identified and then the autoantigen-specific antibody should be evaluated.
2 Platelet-Associated Autoantibodies
van Leeuwen et al. firstly showed that platelet-associated (PA) antibodies eluted from platelets of 42 chronic ITP patients bound to all normal platelets tested, but 32 of these eluates (76%) did not react with platelets obtained from Glanzmann thrombasthenia (GT) [9]. Because GT is a congenital bleeding disorder that is caused by a quantitative defect in platelet membrane glycoprotein (GP)IIb-IIIa [10, 11], the findings of van Leeuwen et al. suggested the importance of GPIIb-IIIa as a major autoantigen in chronic ITP. Varon and Karpatkin showed impaired binding of the monoclonal antibody 3B2, which is specific for GPIIb, to platelets obtained from 15 of 16 chronic ITP patients. Their data also suggested that GPIIb-IIIa may be important as an autoantigen in ITP, and the autoantigen may locate close to the 3B2-binding site because of the possible inhibition of the 3B2 binding by ITP autoantibodies [12]. Subsequently, direct evidence for the presence of anti-GPIIb-IIIa and/or anti-GPIb-IX autoantibodies in ITP has been provided by several different assays including monoclonal antibody-based antigen-specific assays such as the immunobead assay and monoclonal antibody immobilization of platelet antigens (MAIPA) (Fig. 1) [13–15]. Tomiyama et al. developed a direct immunoprecipitation assay: ITP platelets with autoantibodies were directly radiolabeled and solubilized without adding any antibody, and then platelet antigen-antibody complexes were immunoprecipitated by protein A and analyzed by SDS-PAGE. This method allows us to analyze both PA autoantibodies and their target antigens (i.e., autoantigens) simultaneously. Employing this procedure, GPIIb-IIIa and a 56 kd protein were identified as autoantigens for PA autoantibodies in four and three of six ITP patients, respectively, and suggested that the levels of these PA autoantibodies were related to the disease activity (Fig. 2) [16, 17]. Prospective studies revealed that PA autoantibodies were detected in 50–60% ITP patients, and the most frequently detected antibodies were anti-GPIIb-IIIa followed by anti-GPIb-IX antibodies [18, 19]. Although not all but 50–60% ITP possessed PA autoantibodies, the specificity of these autoantibodies for ITP was high (>90%), and the degree of their positivity increased with the severity of ITP [19]. Moreover, corticosteroids and splenectomy resulted in a decrease in PA autoantibody levels associated with an improvement in the platelet count [20]. Taken together, these data indicate that PA autoantibodies play a crucial role in the pathophysiology of ITP.

Fig. 2
Demonstration of platelet-associated (PA) antibodies and their target antigens by a direct immunoprecipitation procedure in ITP. Upper left panel: Procedure for direct immunoprecipitation assay. Lower panel: Direct immunoprecipitation experiments before and after splenectomy are shown. Note that anti-GPIIb-IIIa autoantibodies disappeared after splenectomy. IP immunoprecipitation, IgG immunoglobulin G; MW molecular weight
3 Serum Autoantibodies
Serum (or plasma) antibodies specific for GPIIb-IIIa and/or GPIb-IX have also been demonstrated in ITP by monoclonal antibody-based antigen-specific assays [14]. However, serum autoantibodies were less frequently detected than PA autoantibodies (43.2 vs. 79.7%) [14, 21]. Some serum anti-GPIIb or anti-GPIIIa antibodies could be demonstrated in an immunoblot assay, suggesting that these antibodies might recognize linear structure rather than tertiary structure of GPIIb or GPIIIa [22, 23]. Moreover, some of these serum antibodies showed a different specificity from PA autoantibodies. Fujisawa et al. demonstrated that plasma samples from chronic ITP patients contained antibodies against a cytoplasmic region of GPIIIa and clearly showed the different specificities of PA and plasma autoantibodies to GPIIb-IIIa [24, 25]. Similarly, Kosugi, et al. also demonstrated the different specificity between PA and plasma antibodies specific for GPIIb-IIIa and the presence of anti-αvβ3 antibodies in sera from chronic ITP but not in PA autoantibodies [26, 27]. Moreover, antibodies against vinculin, an intracellular protein, have been detected in ITP sera and control sera. The incidence of antivinculin antibodies in ITP sera (67%; 55 of 82 sera) was significantly higher than that in control sera (40%; 32 of 80 sera), and there is a significant difference between the mean levels of antivinculin antibodies in ITP and control sera [28]. It is possible that antivinculin antibodies may secondarily be induced by platelet destruction in ITP. Taken together, the presence of antibodies against a cytoplasmic region of GPIIIa and/or vinculin in ITP sera as well as the different specificity between PA and serum (plasma) antibodies strongly suggest that sera in chronic ITP may contain antibodies secondarily caused by platelet destruction. Accordingly, in order to reveal the pathophysiology of ITP, we should investigate autoantigens recognized by PA autoantibodies.
4 Characterization of PA Anti-GPIIb-IIIa Autoantibodies
PA anti-GPIIb-IIIa autoantibodies are most frequently detected in ITP [18, 19]. GPIIb and GPIIIa forms a non-covalently associated, divalent cation-dependent heterodimer (αIIbβ3), which is a prototypic integrin and plays a crucial role in normal hemostasis and platelet aggregation as a physiologic receptor for fibrinogen and von Willebrand factor. Dissociation of the GPIIb-IIIa complex into free GPIIb and GPIIIa by EDTA treatment markedly impaired the reactivity of PA anti-GPIIb-IIIa autoantibodies [26, 29]. These findings suggest that most PA autoantibodies recognize cation-dependent conformation(s) of either GPIIb or GPIIIa or conformation(s) formed by both GPIIb and GPIIIa. Bowditch et al. examined the reactivity of PA autoantibodies against five large recombinant GPIIIa peptides. However, they failed to localize the autoantigenic epitopes on GPIIIa [30]. GPIIb-IIIa (αIIbβ3) and αvβ3 share the same β subunit, and GPIIb and αv show strong similarities in primary structure (34% amino acid sequence identity). We took advantage of molecular similarities between these two integrins and investigated whether PA anti-GPIIb-IIIa autoantibodies might also react with αvβ3 expressed on human umbilical vein endothelial cells. We did not detect PA anti-αvβ3 autoantibodies in chronic ITP, and we found that PA anti-GPIIb-IIIa autoantibodies did not recognize αvβ3. Because even topographic epitopes on GPIIIa, such as HPA-1a and HPA-4a, are expressed on both GPIIb-IIIa and αvβ3, these findings suggest that PA autoantibodies are highly GPIIb-IIIa-specific and recognize the tertiary structure of intact GPIIb-IIIa [27].
5 Autoantigenic Epitopes on GPIIb-IIIa
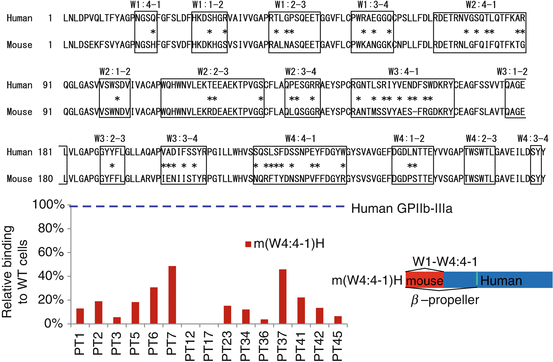
PA anti-GPIIb-IIIa autoantibodies failed to react with αvβ3, suggesting an important role of GPIIb as an autoantigen. Extracellular domain of GPIIb (αIIb) consists of four domains: β-propeller domain, thigh domain, calf-1 domain, and calf-2 domain. The N-terminal globular head of αIIbβ3, which contains a ligand-binding site of this integrin, is formed by a seven-bladed β-propeller domain (W1–W7) from GPIIb and a β I-domain from GPIIIa (β3) (Fig. 3). In addition to the quantitative defect, a qualitative defect in GPIIb-IIIa is responsible for GT phenotype (variant GT) [10, 11]. Molecular characterization of variant GT is informative in defining functionally important site(s) in GPIIb-IIIa, and ligand-binding sites within the N-terminal globular head have been identified in both GPIIb and GPIIIa [11]. By the characterization of a variant GT, CAM, Loftus et al. first demonstrated Asp residue at 119 in GPIIIa as one of the critical residues for ligand binding [31]. Honda et al. demonstrated that a 2-amino acid insertion (Arg-Thr) between amino acid residues 160 and 161 within the GPIIb W3 4-1 loop is responsible for a ligand-binding defect in a variant GT, KO [32]. Both KO and CAM variants GPIIb-IIIa showed markedly impaired ligand binding without disturbing surface expression or inducing major structural change in the receptor. To localize the autoantigenic epitope(s), we examined the reactivity of platelet eluates (PA autoantibodies) against KO mutant or CAM mutant GPIIb-IIIa-expressing 293T cells. Interestingly, 11 of 34 chronic ITP patients (32%) with PA anti-GPIIb-IIIa autoantibodies showed marked decrease in the reactivity with KO GPIIb-IIIa, but not with CAM GPIIb-IIIa [17, 33]. Thus, in one-third of ITP patients, the W3 4-1 loop and its surrounding regions of β-propeller domain in GPIIb may directly or indirectly participate in the formation of autoantigens.

Fig. 3
Structure of extracellular domain of GPIIb-IIIa (integrin αIIbβ3) and β-propeller domain of GPIIb (αIIb). The N-terminal portion of GPIIb, known as the β-propeller domain, contains seven radially arranged β-sheets, termed “W” because of their topology. Each W structure has four antiparallel β-strands and four connecting loops
Autoantigenic epitopes on GPIIb-IIIa were localized on W3 4-1 loop of β-propeller domain in one-third of ITP patients [33]. However, they still remained elusive in the other two-third of ITP patients. In order to further characterize autoantigenic epitopes on GPIIb-IIIa in ITP patients including the remaining two-thirds of ITP patients, Kiyomize et al. systematically examined the reactivity of PA anti-GPIIb-IIIa autoantibodies obtained from 15 ITP patients with human-mouse GPIIb chimeras complexed with human GPIIIa [34]. Firstly, we found that PA anti-GPIIb-IIIa autoantibodies poorly reacted with mouse GPIIb-IIIa mainly due to the difference between human and mouse GPIIb, which is consistent with the previous results [27, 33]. Mouse GPIIb shows higher homology with human GPIIb than human αv (81 vs. 34% amino acid sequence homology), which enabled us to further localize the epitopes. Employing αIIb-αvβ3 chimeras, McMillan et al. demonstrated that 8 of 14 PA anti-GPIIb-IIIa autoantibodies bound to epitopes existing between L1 and Q459 in GPIIb, which correspond to W1–W7 in the β-propeller domain [35]. We narrowed down the autoantigenic epitopes and demonstrated that PA anti-GPIIb-IIIa autoantibodies mainly recognized the N-terminal half of the β-propeller domain (L1-W235 corresponding to W1–W4 4-1 loop) in all 15 ITP patients examined (Fig. 4) [34]. Antigenic loop structures in the β-propeller domain show a significant difference between human and mouse amino acid sequence (Fig. 4). Our systematic examination with human-mouse αIIb chimeras by focusing the loop structures found three main recognition sites for PA anti-GPIIb-IIIa antibodies: (1) a conformational epitope composed of W1:1-2 and W2:3-4 loops (Group A), (2) a region containing the W1:2-3 loop (Group B), and (3) a region containing the W3:4-1 loop (Group C) [34]. It is noteworthy that we further identified some single residues in these loops that were critical for the reactivity of PA autoantibodies obtained from three ITP patients (Pt 17, 23, and 36), suggesting that some PA autoantibodies behaves like monospecific antibodies (Fig. 5) [34]. In addition, we and others demonstrated that many of PA anti-GPIIb-IIIa antibodies showed apparent clonality because of the restricted κ/λ light chain usage [34, 36, 37], which is consistent of the findings that antigenic epitopes for PA anti-GPIIb-IIIa antibodies are limited [34, 35].