Abstract
Megaloblastic anemias are characterized by the presence of megaloblasts in the bone marrow and macrocytes in the blood. In more than 95% of cases, megaloblastic anemia is a result of folate and vitamin B 12 deficiency. Megaloblastic anemia may also result from rare inborn errors of metabolism of folate or vitamin B 12 . In addition, deficiencies of ascorbic acid, tocopherol, and thiamine may be related to megaloblastic anemia. The causes of megaloblastosis are listed. The clinical features, diagnosis, and treatment of cobalamin and folate deficiency are discussed.
Keywords
Megaloblastic anemias, cobalamin deficiency, Schilling test, folate deficiency, intrinsic factor deficiency, Pernicious anemia, Wolfram syndrome, Orotic aciduria, homocysteine, methylmalonic acid
Megaloblastic anemias are characterized by the presence of megaloblasts in the bone marrow and macrocytes in the blood. In more than 95% of cases, megaloblastic anemia is a result of folate and vitamin B 12 deficiency. Megaloblastic anemia may also result from rare inborn errors of metabolism of folate or vitamin B 12 . In addition, deficiencies of ascorbic acid, tocopherol, and thiamine may be related to megaloblastic anemia. The causes of megaloblastosis are listed in Table 7.1 . The clinical features, diagnosis, and treatment of cobalamin and folate deficiency are discussed later in this chapter (see section ‘General clinical features of cobalamin and folate deficiency’).
|
a Associated in some cases with diabetes and sensorineural hearing impairment and in others with the DIDMOAD syndrome. There is wide clinical heterogeneity of this rare disorder. Only the anemia is responsive to high doses of thiamine.
Vitamin B 12 (Cobalamin) Deficiency
Absorption and Metabolism
Dietary vitamin B 12 (Cbl), 1
1 For the purposes of this chapter, vitamin B 12 , cobalamin, and cbl are used interchangeably. Vitamin B 12 contains a metal ion in the form of cobalt and therefore is also known as cobalamin.
acquired mostly from animal sources, including meat and milk, is absorbed in a series of steps that include:- •
Proteolytic release of Cbl from its associated proteins and Cbl binds to haptocorrin, a cobalamin-binding protein, produced by salivary and esophageal glands.
- •
In the duodenum, after exposure to pancreatic proteases, Cbl is released from haptocorrin.
- •
In the proximal ileum Cbl binds to intrinsic factor (IF), a gastric secretory protein, to form the IF–Cbl complex.
- •
Recognition of the IF–Cbl complex by specific receptors on ileal mucosal cells, which is taken into lysosomes where the IF–Cbl complex is released and IF is degraded.
- •
Transport across ileal cells in the presence of calcium ions.
- •
Release into the portal circulation bound to transcobalamin II (TC II)—the serum protein that carries newly absorbed Cbl throughout the body.
Figure 7.1 shows the pathway of cobalamin absorption, transport, and cellular uptake.
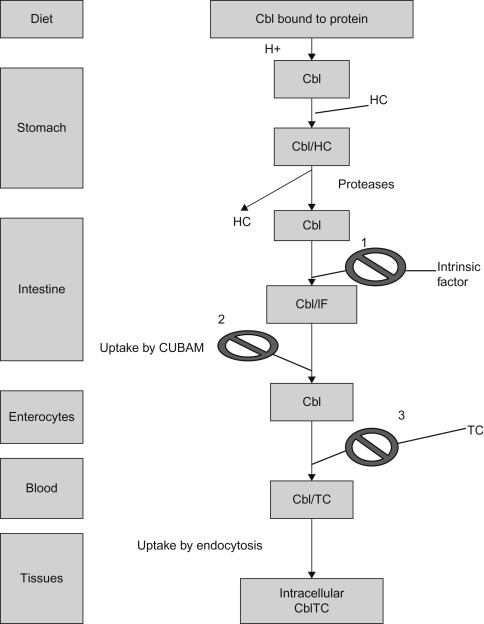
Cobalamin is converted into the two required coenzyme forms, adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl). The cellular metabolism by which the coenzymes are formed involves the following:
- •
Receptor-mediated binding of the TC II–Cbl complex to the cell surface.
- •
Adsorptive endocytosis of the complex.
- •
Intralysosomal degradation of the TC II.
- •
Release of Cbl into cytoplasm.
- •
Enzyme-mediated reduction of the central cobalt atom and cytosolic methylation to form MeCbl or mitochondrial adenosylation to form AdoCbl.
Causes of Vitamin B 12 Deficiency
- •
The causes of vitamin B 12 deficiency are listed in Table 7.2 .
Table 7.2
Causes of Vitamin B 12 Deficiency
- 1.
Inadequate vitamin B 12 intake
- a.
Dietary (<2 mg/day): food fads, lacto-ovo vegetarianism, low animal-source food intake, veganism, malnutrition, poorly controlled phenylketonuria diet
- b.
Maternal deficiency leading to B 12 deficiency in breast milk
- a.
- 2.
Defective vitamin B 12 absorption ( Table 7.3 )
- a.
Failure to secrete intrinsic factor
- i.
Congenital intrinsic factor deficiency (gastric mucosa normal) (OMIM 261000)
- •
Quantitative
- •
Qualitative (biologically inert) a
- •
- ii.
Juvenile pernicious anemia (autoimmune) (gastric atrophy) b
- iii.
Juvenile pernicious anemia (gastric autoantibodies) with autoimmune polyendocrinopathies (OMIM 240300)
- iv.
Juvenile pernicious anemia with IgA deficiency
- v.
Gastric mucosal disease
- •
Chronic gastritis, gastric atrophy (elevated serum gastrin and/or low serum pepsinogen 1 concentrations) often caused by Helicobacter pylori
- •
Corrosives
- •
Gastrectomy (partial/total)
- •
- i.
- b.
Failure of absorption in small intestine
- i.
Specific vitamin B 12 malabsorption
- •
Abnormal intrinsic factor a
- •
Defective cobalamin transport by enterocytes—abnormal ileal uptake (Imerslund–Gräsbeck syndrome) (OMIM 261100)
- •
Ingestion of chelating agents (phytates, EDTA) (binds calcium and interferes with vitamin B 12 absorption)
- •
- ii.
Intestinal disease causing generalized malabsorption, including vitamin B 12 malabsorption:
- •
Intestinal resection (e.g., congenital stenosis, volvulus, trauma)
- •
Crohn’s disease
- •
Tuberculosis of terminal ileum
- •
Lymphosarcoma of terminal ileum
- •
Pancreatic insufficiency c
- •
Zollinger–Ellison syndrome (caused by gastrinoma in duodenum or pancreas)
- •
Celiac disease (gluten enteropathy), tropical sprue
- •
Other less specific malabsorption syndromes
- •
HIV infection
- •
Long-standing medication that decreases gastric acidity (H 2 -receptor antagonists and proton pump inhibitors)
- •
Parasites ( Giardia , Lamblia , Diphyllobothrium latum )
- •
Neonatal necrotizing enterocolitis
- •
- iii.
Competition for vitamin B 12
- •
Small-bowel bacterial overgrowth (e.g., small-bowel diverticulosis, anastomoses and fistulas, blind loops and pouches, multiple strictures, scleroderma, achlorhydria, gastric trichobezoar)
- •
Diphyllobothrium latum , the fish tapeworm (takes up free B 12 and B 12 -intrinsic factor complex), Giardia lamblia , Plasmodium falciparum , Strongyloides stercoralis
- •
- i.
- a.
- 3.
Defective vitamin B 12 transport
- a.
Congenital TC II deficiency (OMIM 275350)
- b.
Transient deficiency of TC II
- c.
Partial deficiency of TC I (haptocorrin deficiency) (OMIM 193090)
- a.
- 4.
Disorders of vitamin B 12 metabolism
- a.
Congenital
- i.
Adenosylcobalamin deficiency CblA (OMIM 251100) and CblB diseases (OMIM 251110)
- ii.
Deficiency of methylmalonyl-CoA mutase (mut, mut 2 )
- iii.
Methylcobalamin deficiency CblE (OMIM 236270) and CblG diseases (OMIM 250940)
- iv.
Combined adenosylcobalamin and methylcobalamin deficiencies: CblC (OMIM 277400), CblD (OMIM 277410), and CblF diseases (OMIM 277380)
- i.
- b.
Acquired
- i.
Liver disease
- ii.
Protein malnutrition (kwashiorkor, marasmus)
- iii.
Drugs associated with impaired absorption and/or utilization of vitamin B 12 (e.g., p -aminosalicylic acid, colchicine, neomycin, ethanol, oral contraceptive agents, Metformin)
- i.
- a.
b Pernicious anemia is the final stage of an autoimmune disorder in which autoantibodies against H + K + -adenosine triphosphatase destroy parietal cells in the stomach.
c Because of lack of the enzymes needed to liberate B 12 from haptocorrin, the protein that initially binds ingested vitamin B 12 . OMIM, Online Mendelian Inheritance in Man ( http://www-ncbi-nlm-nih-gov.easyaccess2.lib.cuhk.edu.hk/omim/ ).
- 1.
Nutritional Deficiency
The recommended dietary allowance of vitamin B 12 for children is 0.9–2.4 μg/day. The most common cause of Cbl deficiency in infants is dietary deficiency in the mother. Mothers following vegetarian, vegan, macrobiotic, and other special diets are at particular risk. Cbl in breast milk parallels that in serum and is deficient when the mother is a vegan or has unrecognized pernicious anemia, has had previous gastric bypass surgery or short gut syndrome.
Defective Absorption
Table 7.3 lists the features of congenital and acquired defects of vitamin B 12 absorption, Table 7.4 lists the main features of genetic defects in processing of vitamin B 12 .
| Stomach | Schilling Test | Serum Antibodies | ||||||
|---|---|---|---|---|---|---|---|---|
| Condition | Histology | Intrinsic factor a | Hydrochloric acid (HCl) | Without IF | With IF | Intrinsic factor | Parietal cell | Associated features |
| Congenital pernicious anemia | Normal | Absent | Normal | Decreased | Normal | Absent | Absent | None; relative of patient may exhibit defective vitamin B 12 malabsorption |
| Juvenile pernicious anemia (autoimmune) | Atrophy | Absent | Achlorhydria | Decreased | Normal | Present (90%) | Present (10%) | Occasional lupus erythematosus, IgA deficiency, moniliasis, endocrinopathy in siblings |
| Juvenile pernicious anemia with polyendocrino- pathies or selective IgA deficiency | Atrophy | Absent | Achlorhydria | Decreased | Normal | Present | Present | Hypothyroidism (chronic auto-immune thyroiditis—Hashimoto’s thyroiditis) insulin-dependent diabetes mellitus, primary ovarian failure, myasthenia gravis, hypoparathyroidism, Addison disease, moniliasis, or selective IgA deficiency |
| Enterocyte vitamin B 12 malabsorption (Imerslund- Gräsbeck) | Normal | Present | Normal | Decreased | Decreased | Absent | Absent | Benign proteinuria, amino-aciduria, no generalized malabsorption b |
| Generalized malabsorption | Normal | Present | Normal | Decreased | Decreased | Absent | Absent | Malabsorption; syndrome; history of ileal resection, Crohn’s disease, lymphoma |
a Either absent secretion of immunologically recognizable IF or secretes immunologically reactive protein that is inactive physiologically. The latter group includes patients whose IF has reduced affinity for the ileal IF receptor, reduced affinity for cobalamin or increased susceptibility for proteolysis.
b Rare cases have been described of this syndrome associated with generalized malabsorption reversed by vitamin B 12 administration and rare cases have been described without proteinuria or aminoaciduria. IF, intrinsic factor.
| Defect | Serum B 12 | Clinical/biochemical |
|---|---|---|
| Food cobalamin malabsorption | Low | N.A.±Anemia, mild ↑ MMA/tHcy |
| Intrinsic factor deficiency | Low | Anemia, delayed development, mild ↑ MMA/tHcy |
| Enterocyte cobalamin malabsorption (Imerslund–Gräsbeck) | Low | Anemia, proteinuria, delayed development, mild ↑ MMA/tHcy |
| Transcobalamin I (R-Binder) deficiency | Low | No abnormality, No ↑ MMA/tHcy |
| Transcobalamin II deficiency | Normal | N.A.±Anemia, failure to thrive, mild ↑ MMA/tHCy |
| Intracellular defects of cobalamin | Normal | Severe disease, ↑ MMA/tHcy |
Gastric Acidity and Peptic Activity Deficiency
Acid pH and peptic activity are required to release cobalamin from the protein-bound state in which it occurs in food. Impaired absorption occurs when there is impaired gastric function, for example atrophic gastritis, partial gastrectomy resulting in a low serum cobalamin, mild increase in methylmalonic acid and homocysteine, and a normal Schilling test.
Intrinsic Factor Deficiency
Patients with absent or defective IF (also known as S-binder) have low serum B 12 and megaloblastic anemia. This autosomal recessive disorder usually appears early in the second year of life, but may be delayed until adolescence or adulthood. The abnormal absorption of cobalamin is corrected by mixing the vitamin with a source of normal IF. Some patients have no detectable IF, whereas others have IF that can be detected immunologically but lacks function. The gene for human IF (GIF gene) has been cloned and localized to chromosome 11. Mutations have been identified in the GIF gene, together with a polymorphism (68A→G) which may be a marker for this inheritance. Homozygous GIF mutations result in complete loss of IF function.
Defective Cobalamin Transport by Ileal Enterocyte Receptors for the Intrinsic Factor-Cobalamin Complex (Imerslund-Gräsbeck Syndrome)
Most of the known patients reside in Norway, Finland, Saudi Arabia, and among Sephardic Jews in Israel. It is due to a selective defect in cobalamin absorption in the ileum that is not corrected by treatment with IF. In these patients, gastric IF level is normal, they do not have antibodies to IF, and the ileal intestinal morphology is normal. In some cases the ileal receptor for IF–cobalamin complex is absent, whereas in other patients it is present. It is an autosomal recessive disorder associated with a low serum B 12 .
The locus for Imerslund–Gräsbeck syndrome has been assigned to chromosome 10. Imerslund–Gräsbeck-causing mutations are found in either of two genes encoding the epithelium proteins: cubilin (CUBN) and amnionless (AMN). The gene receptor, cubilin P1297L (OMIM 602997) 2
2 The six-digit number is the entry number for the disorder in Online Mendelian Inheritance in Man (OMIM) a continuously updated electronic catalog of human genes and genetic disorders. The online version is accessible through the world wide web ( http://www-ncbi-nlm-nih-gov.easyaccess2.lib.cuhk.edu.hk/omim/ ).
is a 640-kDa protein which recognizes IF–cobalamin and various other proteins to be endocytosed in the intestine and kidney. The exact function of AMN is unknown but mutations affecting either of the two proteins may cause Imerslund–Gräsbeck syndrome.The Imerslund–Gräsbeck syndrome usually presents with pallor, weakness, anorexia, failure to thrive, delayed development, recurrent infections, and gastrointestinal symptoms within the first 2 years of life but has been reported up to 15 years of age. In many patients, proteinuria of the tubular type is found that is not corrected by systemic cobalamin.
There has been a decrease in the number of new cases, suggesting that dietary or other factors may influence the expression of this disease.
Defective Transport
Table 7.5 lists clinical manifestations, laboratory finding and treatment of inborn errors of cobalamin transport and metabolism and Figure 7.2 shows the pathways of vitamin B 12 metabolism and sites of inborn errors of vitamin B 12 metabolism.
| Condition (OMIM no.) | Defect | Typical clinical manifestations | Typical onset | Laboratory findings | Treatment and response |
|---|---|---|---|---|---|
| TC II deficiency (OMIM 275350) | Defective/absent TC II | Failure to thrive, megaloblastic anemia, later neurologic features and immunodeficiency | Early infancy 3–5 weeks | Usually normal serum Cbl; elevated serum MMA, homocysteine; absent/defective TC II | High doses of Cbl by injection; good response to treatment if begun early |
| TC I (R-binder) deficiency (OMIM 193090) | Deficiency/absence of TCI in plasma, saliva, leukocytes | Neurologic symptoms (myelopathy) reported, but unclear if these are related to condition | Unclear if observed symptoms are related to condition | Low serum Cbl, normal TC II-Cbl levels. No increase in MMA or homocysteine | Cbl therapy does not appear to be of benefit |
| Defective synthesis of AdoCbl: cblA (OMIM 251100) cblB (OMIM 251110) | Defective synthesis of AdoCbl | Lethargy, failure to thrive, recurrent vomiting, dehydration, hypotonia, keto acidosis hypoglycemia | First weeks or months of life | Normal serum Cbl, homocysteine and methionine; elevated MMA, ketones, glycine, ammonia; leukopenia, thrombocytopenia, anemia | Pharmacologic doses of Cbl, dietary protein restriction, oral antibiotics. Treatment response for cblA better than for cblB |
| Defective synthesis of MeCbl: cblE (OMIM 236270) cblG (OMIM 250940) | Defective synthesis of MeCbl | Vomiting, poor feeding, lethargy, severe neurologic dysfunction, megaloblastic anemia | Most in first 2 years of life | Normal serum Cbl and folate; homocystinuria, hypomethioninemia | Pharmacologic doses of Cbl, betaine; good treatment response in some patients treated early |
| Defective synthesis of AdoCbl and MeCbl: cblC (OMIM 277400) cblD (OMIM 277410) cblF (OMIM 277380) | Impaired synthesis of both AdoCbl and MeCbl | Failure to thrive, developmental delay, neurologic dysfunction, megaloblastic anemia, some cases with retinal findings | Variable from neonatal period to adolescence majority with neonatal onset | Normal serum Cbl, TC II; methylmalonic aciduria, homcystinuria, hypomethioninemia | Pharmacologic doses of hydroxocobalamin, moderate protein restriction, betaine treatment. Response often not optimum |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


